Figure 1: Allosteric MEK inhibitors that reached clinical trials

Sateesh Kumar Vemula1,2* Radhika Katkum3
1College of Medical and Health Sciences, Wollega University, Post Box No: 395, Nekemte, Ethiopia*Corresponding author: Dr. Sateesh Kumar Vemula, Associate Professor, Department of Pharmacy, College of Medical and Health Sciences, Wollega University, Nekemte, Ethiopia, Tel: 057 86 19026; E-mail: vemulasatish15@gmail.com
Development of liquisolid compacts is one of the new pharmaceutical formulation technologies to improve the dissolution rate of poorly soluble drugs. The intent of present investigation was to enhance the dissolution rate of poorly soluble drug flurbiprofen by delivering the drug as a liquisolid compact. Liquisolid compacts were developed using polyethylene glycol 400 (PEG 400) as solvent, Avicel PH102 or starch or HPMC or PEG 4000 or PEG 6000 as the carrier powder and Aerosil 200 as the coating material. The crystallinity of the newly formulated drug and the interaction between excipients was examined by differential scanning calorimetry. The dissolution studies for the liquisolid formulation and the marketed product were carried out at to determine the improvement in dissolution rate and finally the best formulation was subjected to stability studies to assess the drug stability in the formulation. The results of interaction studies showed no change in the crystallinity of the drug and no interaction between excipients. The percentage drug release of flurbiprofen at 10 min (Q10) and dissolution efficiency were increased from 22.81 ± 1.09% and 16.68 for conventional tablet to 91.52 ± 0.78% and 60.17 for the liquisolid tablet F5. The increase in the dissolution rate was also found to be significant compared to the conventional tablet. From the stability studies, the similarity index was found as 84.75 and it is above 50 indicated the stability of drug in the formulation. In conclusion, the liquisolid technique was considered as a promising approach to improve the dissolution of poorly soluble drugs like flurbiprofen.
Crystallinity; Dissolution efficiency; Liquisolid compact; Poorly soluble; Similarity index
Currently pharmaceutical industries deal with several problems and challenges owing to global competition and increasing demand for better products. The oral route is the preferred route of drug administration, but the drug must be in solution form for the absorption to occur through gastro intestinal tract (GIT). In case of poorly soluble drugs dissolution is the rate limiting step in absorption process [1]. In general drugs with low aqueous solubility (lower than 100 µg/ml) show dissolution-limited and incomplete absorption from the gastrointestinal tract of animals and humans [2]. In the drug discovery, 40% of all newly developed drugs are poorly soluble or insoluble in water that led to ineffective absorption and therapeutic failure [3].
Different types of techniques are reported in the literature to improve the dissolution of poorly soluble drugs. These include solid dispersions [4], crystal engineering [5], ball milling [6], complexation [7], selfemulsifying drug delivery systems [8] and use of mesoporous silica carriers [9]. Recently, liquisolid technique [10] has shown promising approach for the dissolution enhancement. Liquisolid systems are described as dry, non-adherent, free-flowing and compressible powder mixtures acquired by conversion of liquid drugs, drug suspensions or drug solution in nonvolatile solvents with chosen carriers and coating materials. In briefly, this technique convert the drug is dissolved in a nonvolatile liquid to dry, free flowing and compressible solid with the aid of carrier and coat materials. Since non-volatile solvents are used to prepare drug solution/suspension, the liquid is not evaporated and drug is carried with in liquid system and is dispersed throughout the final product. Spireas S and Bolton SM gave mathematical formulae to calculate the required quantities of carrier and coating material that are sufficient to convert liquid medication in to a free flowing solid [10]. The liquisolid technique has shown promising results with the drugs like carbamazepine [11], atorvastatin calcium [12], nimesulide [13] and fenofibrate [14].
Flurbiprofen, a phenylalkanoic acid derivative and classified as nonsteroidal anti-inflammatory drugs, which are widely used for the longterm treatment of chronic rheumatic diseases [15,16]. Flurbiprofen is classified as poorly water soluble class II drug and it is primarily intended to treat painful conditions, which requires fast release of drug [17,18]. It is necessary to improve the dissolution rate to improve the bioavailability of the drug. Various techniques like superdisintegrants method, solid dispersions were reported previously. The present study was aspired to enhance the dissolution of flurbiprofen using liquisolid compaction technique.
Flurbiprofen was a gift sample from FDC Limited, Mumbai, India. Avicel PH102, starch, HPMC, PEG 4000, PEG 6000 (carrier powders), Aerosil 200 (coating material) and Crosspovidone were gift samples from Aurobindo Pharmaceuticals Hyderabad, India. All other chemicals used were of analytical grade.
To select the best non-volatile solvent for dissolving flurbiprofen, solubility studies of the flurbiprofen were carried out in three different non-volatile solvents; PEG 400, Tween 80, and Propylene glycol. Saturated solutions were prepared by adding excess drug to the vehicles and shaking on the incubator shaker (Jeiotech, Korea) for 48 h at 25°C ± 1°C. After this period the solutions were filtered through a 0.45 µm Millipore filter, diluted with distilled water and analyzed by double beam UV-Visible spectrophotometer (Systronics, Hyderabad) at a wavelength of 247 nm against blank (blank sample contained the same concentration of specific solvent used without drug).
Binding capacity defined as the capacity of different excipients (carrier material) to hold liquid and behave like dry powder. This was determined by following simple technique. The constant weights of (5 g) of the different powder excipients were taken and non-volatile solvent was added in an increment of 0.01 ml. The mixture was triturated after each addition to help distribution of the liquid throughout the powder particles. The addition of vehicle and the trituration was continued until mortar contents start to look like dry powder [19].
In liquisolid system, the carrier and coating materials can retain only certain amounts of liquid while maintaining acceptable flow and compression properties depending on the excipients ratio used. The excipients ratio R (R = Q/q) of powder is defined as the ratio between the weights of carrier (Q) and coating (q) materials present in the formulation [20]. Preparation of a liquisolid system with an acceptable flowability and compressibility is possible if a maximum liquid on the carrier material is not exceeded. This characteristic amount of liquid is termed the liquid load factor (Lf). The Lf is defined as the weight ratio of the liquid medication (w) and carrier powder (Q) in the system (i.e., Lf = W/Q) [21]. To calculate the loading factor, non-volatile solvent (liquid medication without drug) was added to 10 g of carrier material and blended for 1min. To this coating material was added and triturated.
Liquisolid powder mixtures of different formulations were evaluated for angle of repose, bulk density, tapped density and compressibility index. The fixed funnel method was employed to measure the angle of repose (θ) and it was calculated using the following formula: Tan θ = h/r. In which, θ is the angle of repose, h is the height of the cone and r is radius of the cone base. To measure the angle of repose, a funnel was fixed to a stand so that the lower tip of funnel was 2.5 cm above the surface. A graph paper was placed on a flat surface. The powder blend was allowed to fall freely on the graph paper through the funnel (6.9 cm diameter), till the tip (8 mm diameter) of heap formed just touches the funnel. The radius of heap was noted and from this angle of repose was determined. Angle of repose less than 30°suggests free flowing properties of the material [22].
The bulk density of a liquisolid powder is determined by measuring the volume of a known mass of powder sample that may have been passed through a screen, into a 50 ml graduated cylinder. Tapped densities of powder samples were determined by a tap density apparatus (Intelli, Kshitij Innovations, India). The apparatus was set for 500 tappings for 5 min at stroke height 20 mm, (100 strokes/min). The compressibility index (Carr’s Index) is a measure of the propensity of a powder to be compressed. It is determined from the bulk and tapped densities and is calculated using the following formula: Carr’s Index = [(ρtap - ρb ) / ρtap] / ×100, in which, ρb is bulk density and ρtap is tapped density [22].
The liquisolid compacts were prepared in accordance to method described by Spireas et al. [10]. Flurbiprofen was dissolved in non-volatile solvent i.e., PEG 400 (based on results of solubility test) to prepare the drug solution. The mixture of carrier-coating materials was added to the liquid medication and blended in a porcelain mortar avoiding excessive trituration and particle size reduction. The mixing was done in three stages; first stage the system was mixed slowly to allow uniform distribution of liquid medication. In second stage the mixture was spread as a uniform layer on the surface of the mortar and left standing for few minutes. In the final stage 5% of disintegrant (Crosspovidone) was added to the powder and mixed thoroughly. The final mixture was compressed into tablets with 12 mm round flat punches using 16 station rotary tabletting machines (Cadmach, Ahmedabad, India).
The designed formulations were studied for their physical properties like weight variation, hardness and friability. For estimating weight variation, 20 tablets of each formulation were weighed using an electronic weighing balance (AW 120, Shimadzu Corporation, Japan). The hardness of six tablets was measured using Monsanto tablet hardness tester. Friability was determined on ten tablets in a Roche friabilator (Electrolab, Mumbai, India). For estimation of drug content, ten tablets were crushed, and 100 mg of the powder was accurately weighed and transferred to a 100 ml volumetric flask. Then, prepare the solution to estimate drug content spectrophotometrically at 247 nm.
In vitro dissolution studies were performed for liquisolid compacts, and conventional tablet. The USP paddle method was used for all in vitro dissolution studies. The dissolution was carried out at different pH using different media 0.1 N HCl (pH 1.2). The rate of stirring was 50 rpm. The amount of flurbiprofen was 10 mg in all formulations. The dosage forms were placed in 900 ml of dissolution medium and maintained at 37 ± 0.5°C. At appropriate intervals, 5 ml of samples were taken and filtered through a 0.45 micron filter (Millipore, USA) and analyzed at 247 nm by UV-Visible spectrophotometer. The mean of six determinations was used to calculate the drug release from each of the formulations [23].
Cumulative percent drug release was plotted as a function of time and percent drug release in 10 min (Q10) was calculated. Initial dissolution rate (IDR) was calculated as percentage dissolved of drug over the first 10 min per minute. Dissolution efficiency (DE) was calculated from the area under the dissolution curve at time t (measured using the trapezoidal rule) and expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time. Relative dissolution rate (RDR) is the ratio between amount of drug dissolved from optimized formulation and that dissolved from the conventional formulation at 10 min [24,25].
To study the possible interaction between flurbiprofen and excipients, Differential scanning calorimetry (DSC) study was carried out on pure drug and the optimized formulation and the thermograms were obtained using DSC (Perkin-Elmer, Shelton, U.S). The analyses were performed under nitrogen (nitrogen flow rate 50 ml/min) in order to eliminate oxidative and pyrrolytic effects at a standard heating rate of 15°C/minute over a temperature range of 50°C–350°C.
To evaluate the drug and formulation stability, stability studies were done according to ICH guidelines. Optimized formulation F5 was sealed in aluminum packaging coated inside with polyethylene, and three replicates were kept in the humidity chamber maintained at 40 ± 2°C and 75 ± 5% RH for six months [26]. Samples were collected after six months of storage and analyzed for the drug content and in vitro dissolution rate and they were subjected to statistical analysis using paired t-test to test the significance of difference at 0.05 level of significance [27]. Then to prove the stability of dosage form, the similarity index was calculated between dissolution rates of optimized tablets before and after storage [28].
The solubility of flurbiprofen in PEG-400, Propylene glycol, Tween-80 is given in Table 2. The table shows that the flurbiprofen has highest solubility in PEG-400.
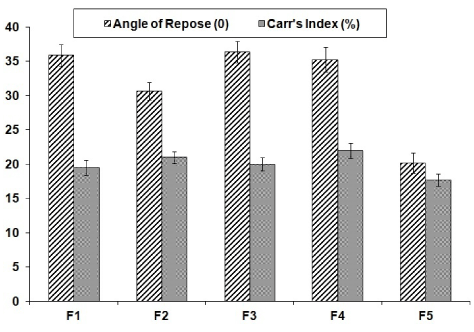
The powder mixtures of different formulations were evaluated for angle of repose and Carr’s index and their values were shown in Figure 1. The results of angle of repose <40 and compressibility index <22 indicates fair to passable flow properties of the powder mixture [22].
The physical properties of flurbiprofen tablets were given in Table 3. In weight variation test, the pharmacopoeial limit for the tablets of not more than 5% of the average weight. The hardness of the tablets was found to be in the range of 3-4 kg/cm2 . Another measure of tablets strength is friability. Conventional compressed tablets that loss less than 1% of their weight are generally considered acceptable. The percentage friability for all formulations was below 1%, indicating that the friability is within the prescribed limits. The tablets were found to contain 95-99% of the labeled amount indicating uniformity of drug content.
The cumulative mean percent of flurbiprofen released from liquisolid compacts containing varying amounts of carrier and coating materials (from F1 to F5) was found to vary from 21.43 ± 0.32% to 91.52 ± 0.78% in first 10 min. This indicates the fast release of drug is observed from above formulations. From the dissolution study, the optimized formulations F5 showed that 99.08% drug release in the first 15 min where as the conventional tablets showed 94.61% in 60 min (Figure 2).
The percent drug release in 10 min (Q10) and initial dissolution rate (IDR) for optimized formulations 91.52 ± 0.78% and 9.15. This is very much higher compared to conventional tablets 22.81 ± 1.09% and 2.28. The improvement in the dissolution characteristics of a drug described in terms of dissolution efficiency (DE) and relative dissolution rate (RDR). The RDR was found to be 4.01 for F5 formulation and the DE was found to be 60.17 for F5 and it is increased much, when compared with the conventional tablet (Table 4).
DSC curves obtained for of pure flurbiprofen and Optimized formulation (F5) were showed in Figure 3. The endothermic peak of flurbiprofen was found 115.6°C and for optimized formulation (F5) was observed 116°C.
To check the stability of tablet formulations, stability studies were carried out for six months. After storage of six months, the formulation was subjected to a drug content and in vitro dissolution studies and from the statistical analysis there was no significant difference between before and after storage (P<0.05). The similarity index value between dissolution profiles of optimized formulation before and after storage was found to be 84.75 (Table 5).
S.No |
Solvent | Solubility mg/ml) |
1 |
PEG 400 | 98.42 ± 3.11 |
2 |
Propylene Glycol | 73.41 ± 2.62 |
3 |
Tween-80 | 61.87 ± 2.39 |
Table 2: Solubility studies of flurbiprofen in non-volatile solvents
Figure 1: Allosteric MEK inhibitors that reached clinical trials
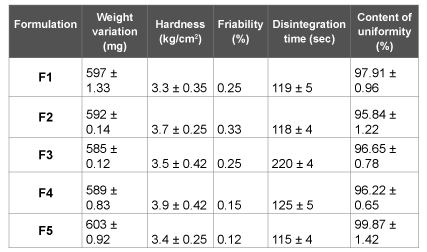
Table 3: Evaluation of flurbiprofen tablets (n=3)
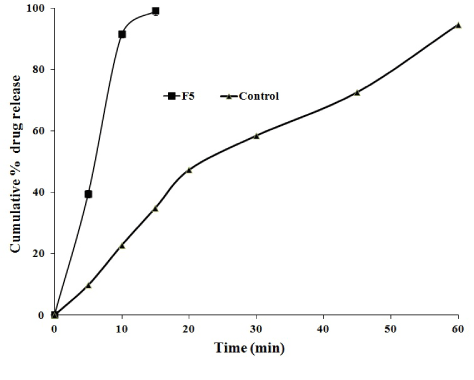
Figure 2: Dissolution profile of flurbiprofen F5 and conventional tablets
| Formulation | (Q10)* | IDR (%/min) | DE | RDR |
| Optimized Formulation (F5) | 91.52 ± 0.78 | 9.15 | 60.17 | 4.01 |
| Conventional tablet | 22.81 ± 1.09 | 2.28 | 16.68 |
Table 4: Dissolution Parameters of flurbiprofen tablets (F5) and conventional tablets (n=3)

Figure 3: DSC thermograms of A) Flurbiprofen B) PEG 6000 C) Mixture of Flurbiprofen and PEG 6000
Flurbiprofen, an NSAID, has a poor water solubility that limits the oral absorption and bioavailability. Hence the objective of the present work was to improve the dissolution rate which in turn would increase the absorption. In the present study flurbiprofen liquisolid tablets were prepared by using PEG 400 as a non-volatile liquid vehicle Avicel PH102 or starch or HPMC or PEG 4000 or PEG 6000 as the carrier powder and Aerosil 200 as the coating material in different ratios and they were characterized for different physical parameters and drug release studies to find the optimized formulation that shows fast dissolution rate (Table 1).
Drug solubility in non-volatile vehicle is most important aspect in formulation of liquisolid systems. The solubility of the drug contributes to formation of molecular dispersion in non-volatile solvent which will improve the dissolution rate. Based on the solubility data (Table 2), PEG 400 was selected as the vehicle for flurbiprofen. The optimum amount of PEG 400 incorporated in to the formulation depends upon the acceptable flow and compressibility of the powder material. The results of angle of repose (<40) and Carr’s index (<22) indicates fair to passable flow properties of the powder mixture. The angle of repose less than 20°indicates good flow, 20°-40° represents reasonable (fair) flow while if values are greater than 40° indicates poor flow [22].
The prepared tablets were studied for their physical properties like weight variation, hardness, friability and drug content uniformity they were complied with pharmacopoeial limits. The tablets should have sufficient hardness to resist the breakage during handling and at the same time it should disintegrate after swallowing. The average percentage deviation of all tablet formulations was found to be within the above mentioned limit and hence all formulations passed the uniformity of weight as per official requirements (Indian Pharmacopoeia., 1996). From the physical characterization of all tablet formulations were uniform in hardness, friability and drug content uniformity.
From the in vitro drug release studies, formulations F5 was considered better among other formulations to produce fast release of the flurbiprofen when compared to other formulations. From the calculations of DE and RDR, F5 formulation showed better improvement in dissolution and it is considered as optimized formulation. Overall increase in the dissolution performance of the optimized formulation was described in terms of dissolution parameters (IDR, DE, RDR) and compared with conventional tablet. DE of flurbiprofen was increased by four times when compared with conventional tablet.
DSC studies showed that there is no interaction between the drug and excipients. The thermograms of the optimized formulation did not show any significant shift in the endothermic peak, indicating that there was no physical change in drug in the liquisolid compacts. After storage of six months, the formulation was subjected to a drug assay and in vitro dissolution studies and the data showed that there was no significant change in formulation in the sense of drug content and dissolution behavior. The similarity index value was found as 84.75, which is more than 50 indicates similarity between the dissolution profile before and after storage [29,30].
The increased dissolution from liquisolid formulation could be due to, presence of drug in solubilised state in the formulation, which contributes to the increased wetting properties, thereby enhancing the dissolution rate. Similarly, as the formulation disintegrates in dissolution media, the drug will be presented in a state of molecular dispersion. This will increase the effective surface area of the particles available for dissolution. In conclusion, development of the liquisolid compacts can be a promising alternative technique for waterinsoluble drugs to achieve the fast dissolution rate.

Table 1: Formulation of flurbiprofen liquisolid tablets

Table 5: Stability studies of flurbiprofen F5 tablets (n=3)
The Liquisolid technique was found to be a promising approach for improving the dissolution rate of poorly soluble drug like flurbiprofen. The dissolution of flurbiprofen was significantly increased in liquisolid formulation compared to conventional tablets. The DE of flurbiprofen was increased by four times in optimized liquisolid formulation F5 when compared with conventional tablet. DSC studies indicate there were no interactions between drug and excipients. From the stability studies, thesimilarity index was found as above 50 indicated the stability of drug in the formulation. The increased dissolution rate may be due to increased wetting and increased surface area of the particles. Thus the liquisolid technique can be a promising approach to improve the dissolution rate of poorly soluble drugs.
The authors acknowledge FDC Limited, Mumbai, India and Aurobindo Pharmaceuticals Hyderabad, India for gift samples. The authors also thank the management of Chaitanya College of Pharmaceutical Sciences for providing facilities.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation: Vemula SK, Radhika K (2015) Liquisolid Compact Technique for Improvement of the Dissolution Rate of Flurbiprofen: Formulation and Evaluation. J Drug Res Dev 1(1): doi http://dx.doi.org/10.16966/2470-1009.102
Copyright: © 2015 Vemula SK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access