Abstract
Background: Pharmacoresistant epilepsy represents 30-40% of cases with intractable epilepsy in children, and up to 20% of adult cases. Various cerebral lesions can lead to drug-resistant focal epilepsy. They are medically refractory but the complete surgical resection correlates with good outcome.
Objective: The purpose of our morphological study is to identify the histopathological findings, which can be observed in patients with pharmacoresistant focal epilepsy.
Methods: This retrospective study included 205 histologically proven cases, diagnosed during the period from 2009 to 2021 in the Epilepsy Surgery Center of University Hospital “Saint Ivan Rilski”, Sofia, Bulgaria. Brain tissue materials were embedded in paraffin using routine histological practice. Tissue sections were deparaffinized and stained with haematoxyllin and eosin. Further histo- and immunohistochemical examinations were performed.
Results: The most common pathomorphological findings were: cortical dyslamination adjacent to a glial or glioneuronal tumors (n=54), isolated forms of focal cortical dysplasia (n=51), cortical dyslamination adjacent to vascular malformations (n=50), hippocampal sclerosis (n=35), cortical lamination abnormalities adjacent to any other lesion acquired during early life (n=14).
Conclusion: The most frequent findings in pediatric group patients with pharmacoresistant focal epilepsy were glioneuronal tumors. The most common epilepsy associated tumors were gangliogliomas and dysembryoplastic neuroepithelial tumors. A single case of an anaplastic ganglioma was described in more detail. The leading cause of pharmacoresistant epilepsy in adult patients was hippocampal sclerosis.
Keywords
Pharmacoresistant focal epilepsy; Cortical dyslamination; Focal cortical dysplasia; Hippocampal sclerosis
Introduction
Focal Cortical Dysplasia (FCD) is a heterogeneous group of conditions that can predispose to the development of pharmacoresistant epilepsy [1]. Traditionally, the term “dysplasia” has been used to refer to precancerous conditions (epithelial dysplasia of colonic, gastric, etc. epithelium). It is sometimes used to refer to disorders of abnormal development, abnormalities in the size, shape and structural organization of mature cells. Blumcke I, et al. [2] define as dysplastic any tissue that is not properly developed during embryonic or fetal life. FCDs include a variety of disturbances in normal brain development: cortical dyslamination, abnormal histoarchitectonics, and alterations of the underlying white matter (e.g., neuronal heterotopia) [2,3].
With the enrichment of knowledge in terms of clinical presentation, pathomorphological findings, radiological and genetic studies in order to better understand FCD, the International League Against Epilepsy (ILAE) Working Group created current three-level classification system in 2011 [2].
The three-tiered ILAE classification system of FCD distinguishes isolated forms (FCD Type I and II) from those associated with another principal lesion, i.e. hippocampal sclerosis (FCD IIIa), tumors (FCD IIIb), vascular malformations (FCD IIIc), or lesions acquired in early life (i.e. traumatic injury or encephalitis, FCD IIId) [2].
Materials and Methods
The present study included patients with focal seizures causing significant negative effects on quality of life, despite treatment with at least two antiepilectic drugs for more than 1 year. The cause of the pharmacoresistant epilepsy had to be a structural lesion according to the type of FCD that was histologically proven after the surgical intervention.
Our study included 205 histologically proven cases, diagnosed during the period from 2009 to 2021 in the Epilepsy Surgery Center of University Hospital “Saint Ivan Rilski”, Sofia, Bulgaria. Brain tissue materials were embedded in paraffin using routine histological practice. Tissue sections were deparaffinized and stained with haematoxyllin and eosin. Further histo-and immunohistochemical examinations were performed in the cases where it was necessary.
One hundred and nine (53.17%) of the patients in the series were male and 96 (46.83%) were female. The mean age at diagnosis was 18.7 years. The minimum age was 1 year and the oldest patient was 57 years old.
We strictly adhered to the International League Against Epilepsy classification in the pathomorphological definition of the different types of FCD [2].
Results
Two hundred and five patients with drug-resistant epilepsy underwent neurosurgery and were pathomorphologically studied between 2009 and 2021. The most common pathomorphological findings were: isolated forms of FCD (n=51)-FCD type I and FCD type II, hippocampal sclerosis (n=35)-FCD type IIIa-HS, gangliogliomas (n=19)-GG, dysembryoplastic neuroepithelial tumors (n=14)-DNT, other glial and glioneuronal tumors (n=21); vascular malformations (n=50); glial scars/gliosis (n=10), encephalitis (n=4); other findings (n=1) (Table 1).
| FCD-type |
Number of patients |
Average age |
Male/female ratio |
| Ia |
3 |
9,3 years |
0:3 |
| Ib |
10 |
19,3 years |
2:8 |
| Ic |
6 |
9,5 years |
3:3 |
| IIa |
26 |
19,3 years |
12:14 |
| IIb |
6 |
13,8 years |
3:3 |
| IIIa |
35 |
33,6 years |
15:20 |
| IIIb |
54 |
22,6 years |
35:19 |
| IIIc |
50 |
21,6 years |
29:21 |
| IIId |
13 |
29,4 years |
9:4 |
Table 1: The distribution of different subtypes of FCD.
In the pediatric group of patients-94 cases, the following distribution of FCD types was observed: the most frequent was FCD type IIIb-29 cases, followed by FCD type IIIc-23 cases. The third most frequent type of FCD was type IIa-16 cases. The remaining patients were distributed as follows: FCD Ia-3, FCD Ib-6, FCD Ic-5, FCD IIb-6, FCD-IIIa-1, FCD-IIId-4, others-1 case.
In adult patients-111 cases the distribution was as follows: the most frequent is type IIIa FCD-35 cases, followed by type IIIc FCD-27 cases. The third most frequent type of FCD was type IIIb-25 cases. The remaining patients were distributed as follows: FCD Ia-0, FCD Ib-4, FCD Ic-1, FCD IIa-10, FCD IIb-0, and FCD-IIId-9.
The total number of tumors was 54, 15 of which were low grade glial tumors-WHO grade I and II: 7 pilocytic astrocytomas, 4 oligodendrogliomas, 4 others. The youngest patient was 7 years old with oligodendroglioma, WHO grade II, and the oldest patient was 51 years old, also diagnosed with oligodendroglioma, WHO grade II. The primary goal in this group of low-grade glial tumors was removal of the tumor mass in conjunction with saving or prolonging the patient’s life. They showed some evidence of cortical dyslamination.
The mean age in the type IIIb FCD was 24.9 years. Thirty-nine patients were male and 16 were female.
Of particular interest were 39 cases of tumors, including 19 GG, 14 DNT, 2 pleomorphic xanthoastrocytomas, 2 rossette forming glioneuronal tumors, 1 subependymal giant cell astrcytoma, and 1 hypothalamic hamartoma.
Discussion
The diverse spectrum of epileptogenic lesions in cases with FCD includes isolated forms (FCD type I and type II) as well as those associated with other epileptogenic lesions (FCD type III). Although the pathomorphologic characteristics of the individual forms are well known, their etiologic and pathogenetic features remain incompletely understood.
FCD type I is an isolated form characterized by a histoarchitectural disruption (dyslamination) of the six-layered structure of the cerebral cortex. It affects one or more lobes and is found in young patients with severe epilepsy and psychomotor retardation [4-6]. According to the type of changes, three subtypes can be distinguished: Ia with radial dyslamination and microcolumn formation (Figure 1A), Ib with tangential (horizontal) presented dyslamination, and a combination of both, known as type Ic FCD [2]. In our patient series, FCD type I was present in 19 cases. The mean age of diagnosis was 12.7 years. The predominant sex was female.

Figure 1: Isolated forms FCD: A-type Ia-radial dyslamination with microcolumns formation (Hematoxylin and eosin stain, x100); B-type IIa-cortical dyslamination with presence of dysmorphic neurons (HE, x200); C-type IIb-cortical dyslamination with presence of balloon cells (Hematoxylin and eosin stain, x200); D-type IIb CD34-positive balloon cells (IHC, x200).
FCD type II includes two subtypes: FCD type IIa-cortical dyslamination with the presence of dysmorphic neurons without balloon cells; FCD type IIb-cortical dyslamination with dysmorphic neurons and balloon cells (Figures 1B and 1C). The manifestations of cortical dyslamination of FCD type II are marked-with indistinguishable layers of the cerebral cortex, except for the molecular layer (lamina molecularis). In addition, there is obliteration of the cerebral graywhite matter boundary, with frequent heterotopic neurons appearing in the white matter. The balloon cells are demonstrated by additional immunohistochemical studies with vimentin and GFAP-δ [7-9]. Some ballooned cells express CD34 (Figure 1D). We observed 32 cases (26- with dysmorphic neurons and 6-with balloon cells) of FCD type II. The mean age at diagnosis was 16.6 years, with a relatively equal sex distribution.
According to literature data HS is the most common pathomorphological finding in adult patients with pharmacoresistant temporal lobe epilepsy [10-14]. In a series of 9523 epilepsy patients in Europe, HS was diagnosed in 36.4% after surgical treatment, with 5% of them demonstrating so-called dual pathology-a combination of HS with cortical malformations (e.g., FCD type IIa and FCD type IIb), epileptogenic tumors, vascular lesions, or inflammatory manifestations [14].
Histologically, HS is characterized by areas of pyramidal cell loss in CA1 (Zommer’s sector), CA3 and CA4, while CA2 pyramidal cells and granular cells of the gyrus dentatus remain relatively intact (Figures 2A-2C). Hippocampal sclerosis accounted for 17.1% of our patients. It is noteworthy that 94 of all 205 patients were under 18 years of age. The mean age at diagnosis of HS was 33.6 years. In addition to the wellknown morphological changes in our cases, the increased number of string blood vessels was striking (Figure 2D). Dual pathology-type IIIa FCD HS combined with one or more lesions other than type I FCD.
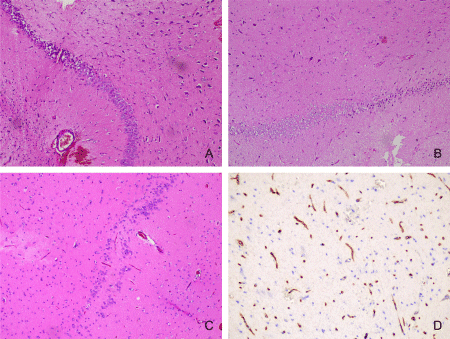
Figure 2: FCD type IIIa-HS: A, B-loss of neurons and gliosis predominantly in CA4 and CA1 fields (Hematoxylin and eosin stain, x100); C-neuronal loss and bilamination in CA4 field (Hematoxylin and eosin stain, x100); D-Collagen IV-positive “string” blood vessels in HS (Immunohistochemical staining for Type IV collagen, x200).
In 10 (8 males and 2 females) of 35 cases with HS we had dual pathology-with malformative leptomeningeal vessels presented in 8 and dysmorphic neurons in the lateral temporal cortex in 2 cases.
The long-term epilepsy-associated tumors are often low-grade tumors with a predominant glial component (astrocytomas) or, most frequently, mixed dysplastic neuronal and neoplastically transformed glial elements (GG and DNT) [15]. The most frequent tumor associated with FCD was GG, with a total of 19 cases, representing 9.27% of the entire series of 205 cases, and 25.93% of cases of FCD IIIb. Seventeen of the GGs had conventional morphology (Figure 3A), 1 was desmoplastic, in a 4-year-old boy, and 1 showed features of anaplasia-malignant transformation-in a 4-year-old girl. The mean age of diagnosis of GG was 17.1 years. The minimum age here was 2 years, and the oldest patient was 44 years old. Of particular interest to us was the case of glioneuronal tumor in a 4-year-old child, located in the temporal lobe, clinically manifested with pharmacoresistant epilepsy. The histological examination of surgically removed formation showed proliferation of cells with relatively monomorphic, round nuclei with coarse chromatin and eosinophilic cytoplasm. Single neuronlike cells with vesicular nuclei, prominent nucleoli, and eosinophilic cytoplasm with processes were found among the tumor cells. There were small areas, where tumor was sharply separated from the cerebral cortex. Neoplastic cells with rhabdoid morphology and intranuclear pseudoinclusions were seen. Reticular fibers were single, mostly perivascular located. Further immunohistochemical studies showed positive reaction in neoplastic cells for GFAP and Olig-2. Tumor nuclei expressed INI1. The single neuron-like cells were immunoreactive with MAP2, neurofilament and synaptophysin. The proliferative activity reached 20%. The diagnosis of an anaplastic ganglioglioma was made (Figures 4A-4D). DNT was the second most common tumor, with a total of 14 cases, representing 6.83% of all cases of FCD in our series of patients and 25.93% of FCD-associated tumors. The majority of cases belonged to the simple “classical” histological forms of DNT, and 2 patients demonstrated complex morphology-a combination between classical and the presence of so-called glial nodules (Figure 3B). The mean age at diagnosis of DNT was 23.14 years. The youngest patient was 9 years old, the oldest was 44 years old. The spectrum of tumors associated with FCD was expanded by 2 cases of pleomorphic xanthoastrocytomas in two boys of the same adolescent age-18 years (Figure 3C), 2 rossette forming glioneuronal tumors in a 19-year-old female and in a 27-year-old male (Figure 3D), a case of subpendymal giant cell astrocytoma and hypothalamic hamartoma diagnosed in a 14-and 17-year-old girl, respectively
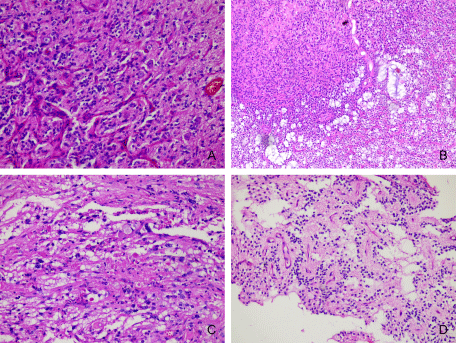
Figure 3: FCD IIIb: A-GG-glioneuronal tumor with mild to moderate cellularity and presence of dysplastic neurons in the midst of fibrillary glial proliferation (Hematoxylin and eosin stain, x200); B-DNT-glioneuronal tumor with the presence of glioneuronal complexes, microcystic spaces with mucinous content and a glial nodule, demonstrating the complex characteristics of the tumor (Hematoxylin and eosin stain, x100); C-pleomorphic xanthoastrocytoma with scattered eosinophilic granular bodies (Hematoxylin and eosin stain, x200); D-rossette forming glioneuronal tumor with neurocytic rosettes and perivascular pseudorosettes (Hematoxylin and eosin stain, x100).
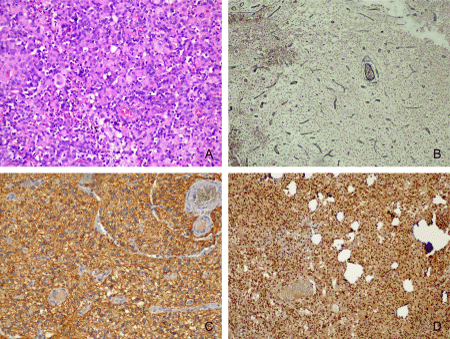
Figure 4: Anaplastic ganglioglioma: A-neoplastic cells with rhabdoid morphology and intranuclear pseudoinclusions (Hematoxylin and eosin stain, x200); B-perivascular located reticular fibers (Reticulin stain, x100); C-positive reaction in neoplastic cells for GFAP (Immunohistochemical staining for Type IV collagen for Glial fibrillary acidic protein, x200); D-preserved expression for INI1, which excludes atypical teratoid/rhabdoid tumor (Immunohistochemical staining for Integrase interactor 1, x100).
It has been suggested that vascular malformations represent congenital lesions determined by disruption of mesodermal differentiation [16]. Type IIIc FCDs included capillary haemangiomatosis, meningoangiomatosis (Figure 5A), cavernous and arterio-venous malformations (Figure 5B), leptomeningeal angiomatosis (e.g. Sturge-Weber syndrome-Figure 5C). Leptomeningeal vascular malformations were one of the most common epileptogenic lesions in our patients, both in the paediatric and adult groups.
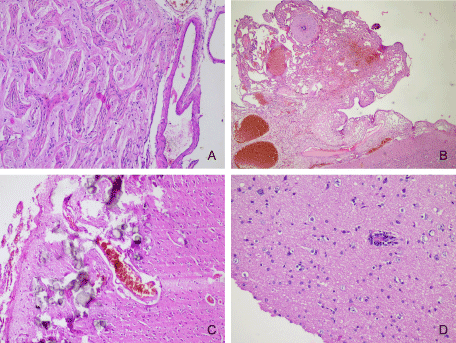
Figure 5: A-FCD type IIIc-meningioangiomatosis with leptomeningeal and meningovascular proliferation (Hematoxylin and eosin stain, x100), B-FCD type IIIc-malformative leptomeningeal blood vessels (Hematoxylin and eosin stain, x40); C-FCD type IIIc-leptomeningeal angiomatosis with cortical microcalcifications in a case with Sturge-Weber syndrome (Hematoxylin and eosin stain, x100); D-FCD type IIId-perivascular lymphocyte infiltration-Rasmussen’s encephalitis (Hematoxylin and eosin stain, x200).
Areas of gliosis are less frequently found to be epileptogenic-10 cases in our series. Epileptic epileptic seizures are frequently seen in patients with encephalitis [17]. Cytotoxic T lymphocytes and antibodymediated complement activation are the most important components of the adaptive immune system that could induce neurodegenerative changes that eventually lead to epileptic encephalitis [18]. On the other hand, the innate immune system acts through interleukin-1 and Toll-like receptor-associated mechanisms playing a direct role in epileptogenesis [19,20].
We diagnosed 4 cases of encephalitis in our patients, including one case of Rasmussen’s encephalitis (Figure 5D).
Double pathology is a combination between different types of FCD, differing from FCD type I and FCD type IIIa.
Double pathology was present in only 3 cases, a 12-year-old boy with a combination of balloon cells and malformative leptomeningeal blood vessels and a combination between GG and leptomeningeal vascular malformation in a 3-year-old boy and in a 33-year-old man.
Conclusion
Surgical methods used in order to control epileptic seizures are varied, with the main type of surgical intervention being the removal of the epileptogenic zone. Only the subsequent histological examination of the materials enables a definitive diagnosis to be made for the various epileptogenic lesions.
The most frequent findings in pediatric group patients with pharmacoresistant focal epilepsy were long-standing epilepsyassociated tumors, followed by cortical dyslamination combined with vascular malformation. Isolated Forms of Cortical Dyslamination (FCD type I and II) were more common here.
The adult group of patients was dominated by cases with FCD type IIIa-HS, which may be associated with vascular malformations in the context of the so-called dual pathology.
The majority of long-standing epilepsy-associated tumors are benign, low-grade. Malignant transformation may be observed rarely. The cerebral cortex demonstrates features of cortical dyslamination adjacent to the GG, DNT and other rarer epileptogenic tumors.
