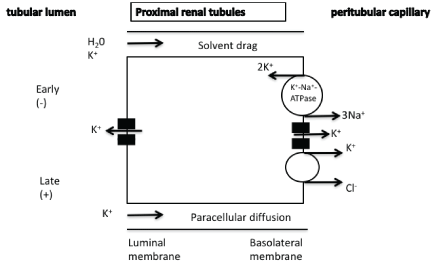
Figure 1: Passive reabsorption and paracellular diffusion of potassium in the proximal renal tubules.
Liontos A Filippatos TD Elisaf MS* Liamis G
Department of Internal Medicine, Medical School, University of Ioannina, Ioannina, Greece*Corresponding author: Moses S Elisaf, Department of Internal Medicine, Medical School, University of Ioannina, Ioannina, Greece, Tel: +30 2651007509; Fax: +30 2651007016; E-mail: melisaf54@gmail.com
Potassium homeostasis, which is an essential part of all metabolic functions, is primarily modulated by renal mechanisms. The aim of this short review is the detailed presentation of the renal mechanisms involved in potassium homeostasis. Proximal renal tubules reabsorb the bulk of filtered potassium via the paracellular pathway, while in the thick ascending limb of Henle potassium reabsorption occurs via the Na+/K+/2Clcotransporter. Accordingly, increased serum potassium levels induce decreased sodium reabsorption in the proximal renal tubules, in the thick ascending limb of Henle and in the early distal convoluted tubules (DCT1) resulting in increased sodium and tubular flow in the distal part of the nephron which facilitates potassium secretion. The final potassium excretion in the distal part of the nephron is dependent on the underlying potassium homeostasis. In individuals with a positive potassium balance, potassium secretion is observed in the distal nephron through the renal outer medullary potassium (ROMK) and maxi-K+ channels. Aldosterone plays a fundamental role in this distal potassium secretion. Furthermore, increased sodium and tubular flow in the distal nephron and the increased electronegativity of the lumen can substantially increase potassium secretion in the distal nephron. In individuals with a negative potassium balance, potassium reabsorption is observed in the alpha-intercalated cells of the collecting tubules through H+/K+-ATPase . Beyond the feedback renal control of potassium homeostasis, there is also a feedforward control involving gut hormones or other unidentified mechanisms. It is worth mentioning that magnesium homeostasis affects renal potassium secretion since hypomagnesaemia is followed by up regulated ROMK channels and kaliuria. In conclusion, multiple renal mechanisms are involved in potassium homeostasis.
Potassium; Kidney; Renal Tubules; Kaliuria; Aldosterone; Magnesium
Renal potassium homeostasis plays a key role in the overall potassium balance [1-3]. In the following short review, the most important mechanisms affecting potassium homeostasis are analyzed, focusing on recently published data regarding renal mechanisms.
There are two general mechanisms involved in renal potassium homeostasis
a) the Feedback control related to hormones affecting potassium homeostasis, such as insulin that promotes potassium entry into cells through the Na+/K+-ATPase and aldosterone that increases potassium excretion, and
b) the Feedforward control which is modulated by the gut through not well-defined mechanisms affecting the activity of channels and transporters that regulate renal potassium handling.
Physiologically, the bulk of filtered potassium (60-70%) is reabsorbed in the proximal renal tubules predominantly via the paracellular pathway. There are experimental studies suggesting that there may be a small component of transcellular potassium transport. The underlying mechanisms of potassium reabsorption include a solvent drag mechanism, the difference of potassium concentration between lumen and cell (potassium concentration is higher in the lumen upon fluid absorption) as well as the favorable electrochemical difference especially in the late part of the tubule when the luminal voltage shifts to slightly positive, favoring paracellular potassium diffusion. In the basolateral membrane, potassium can exit the cell through potassium channels or through K+/ Cl−cotransportation (Figure 1) [1-4].
Figure 1: Passive reabsorption and paracellular diffusion of potassium in the proximal renal tubules.
It has been suggested that in cases of increased serum potassium levels the resultant elevated peritubular potassium can decrease transcellular potassium reabsorption in the proximal tubule resulting in increased sodium and tubular flow to the more distal parts of the nephron facilitating potassium secretion [5,6]. A recently published experimental study showed that the hyperkalemia-induced depolarization of the peritubular membrane is associated with chloride and bicarbonate anions retention into the tubular cells (reduced sodium-bicarbonate co-transportation) [6]. The resultant alkalization of the cell is associated with reduced sodium-hydrogen exchanger activity and decreased sodium reabsorption as the carrier promotes both sodium reabsorption and hydrogen secretion into the lumen[5,6]. The opposite mechanisms are implicated in case of decreased serum potassium levels.
In the thick ascending limb of Henle potassium reabsorption and excretion occurs both paracellularly (as in the proximal tubules) but also through transcellular mechanisms via the apically located Na+/K+/2Cl- - cotransporter (NKCC2) and the apically located renal outer medullary potassium (ROMK) channels. Basolateral Na+/K+-ATPase leads to low intracellular sodium which provides the favorable gradient for the activity of Na+/K+/2Cl- -cotransporter. The ROMK channels recycle potassium into the lumen and
a) result in lumen positive voltage, which favors the passive paracellular potassium reabsorption and
b) ensure an adequate amount of potassium in the lumen to sustain co-transporter activity.
Potassium entering the cell can exit the cell through potassium channels or in co-transport with chloride or bicarbonate anions (Figure 2) [1-4].
It has recently been reported that in the medullary thick ascending limb of Henle hyperkalemia-associated increased interstitial potassium (increased paracellular potassium reabsorption due to enhanced medullary recycling) can also decrease sodium-chloride reabsorption through the NKCC2. It has been suggested that potassium-associated depolarization of the peritubular membrane is associated with dephosphorylation and down-regulation of NKCC2 leading to increased sodium and tubular flow in the distal part of the nephron and facilitating potassium secretion [5,6]. The opposite mechanisms are implicated in case of decreased serum potassium levels.
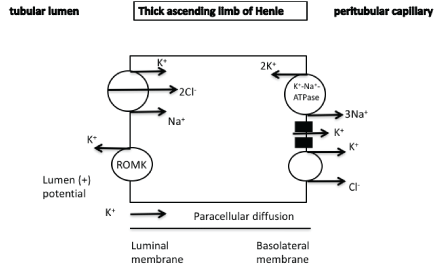
Figure 2: Paracellular diffusion and transcellular reabsorption of potassium in the thick ascending limb of Henle.
K+/Na+/2Cl --cotransporter
ROMK channel : renal outer medullary potassium channel
Although only 5-10% of the filtered potassium reaches the distal nephron, its final balance is regulated by this segment. The final potassium excretion in the urine is dependent on underlying potassium levels.
i) In cases of a positive potassium balance, potassium secretion in the distal nephron is observed in the distal convoluted tubules (DCT), in the connecting tubules (CNT) and in the cortical collecting tubules (CCT) (mainly in the principal cells).
The DCT can be divided into early and late segments (DCT1 and DCT2, respectively). Both of these segments express the thiazide-sensitive Na+/Cl--cotransporter (NCC), an electroneutral cotransporter that is structurally similar to NKCC2. Potassium secretion (through the ROMK channels) in the DCT1 is mediated via sodium chloride reabsorption by the apically located thiazide-sensitive NCC (Figure 3). The Na+/K+-ATPase-mediated low cellular sodium concentration provides a favorable sodium gradient for sodium reabsorption as it enhances the activity of the NCC [1-4]. The reabsorption of cationic sodium makes the lumen electronegative, thereby creating a favorable gradient for the secretion of potassium into the lumen via potassium channels in the apical membrane. It has been recently suggested that the DCT1 plays a prominent role in potassium secretion in cases of changes of dietary potassium intake. It seems that small increases in potassium concentration after potassium intake is followed by a decreased activity of NCC due to its dephosphorylation resulting in increased sodium and flow delivery to the DCT2, CNT, and CCT that are the aldosterone-sensitive potassium secretory segments (see below for details) [1-4]. In the late distal convoluted tubules as well as in the connecting tubules and in the principal cells of cortical collecting tubules, potassium secretion is mediated by both ROMK and maxi-K+ channels [also known as the large conductance potassium channels (BK)]. An electroneutral K+/Cl- -cotransporter is also present and contributes to potassium secretion (Figures 3 and 4). Thus, in cases of low luminal chloride concentration, increased potassium secretion is observed through this cotransporter [1]. The Na+/K+-ATPase located in the basolateral cellular membrane is responsible for the active transport of potassium into the cells leading to elevated cellular potassium concentration and provides a favorable diffusion gradient for potassium movement into the lumen. Similarly, the Na+/K+-ATPase-mediated decreased sodium concentration in the cells can maintain a favorable diffusion gradient for sodium reabsorption
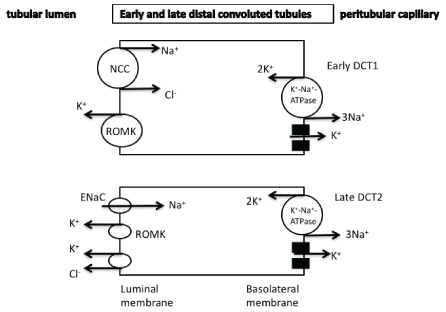
Figure 3: Potassium secretion is observed in the distal convoluted tubules mainly through the ROMK channels. In the late DCT2 aldosterone affects potassium secretion.
NCC: thiazide-sensitive Na+/Cl- -cotransporter
ROMK: renal outer medullary potassium channel
DCT: distal convoluted tubules
ENaC : epithethial sodium channel
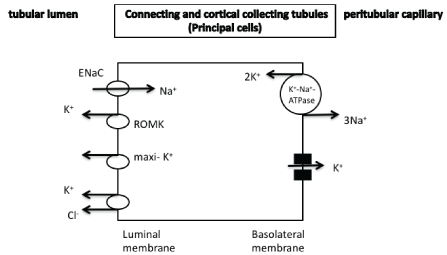
Figure 4: Potassium secretion in the principal cells of collecting tubules through both ROMK and maxi-K+ potassium channels. Aldosterone affects potassium secretion, which is also modulated by the difference in potassium concentration and also by the difference in electrical gradient between cells and lumen. An electroneutral K+/Cl--cotransporter is also observed.
ROMK: renal outer medullary potassium channel
maxi-K+ potassium channels: Large-conductance calcium-activated
(maxi-K+, BK) potassium channels
ENaC : epithethial sodium channel
In the distal nephron (DCT2, CNT, CCT) aldosterone plays a cardinal role in potassium excretion since it can
a) increase cellular potassium concentration by the stimulation of the basolateral Na+/K+-ATPase
b) increase sodium reabsorption through the amiloride-sensitive epithelial sodium channel (ENaC) leading to electronegativity of the lumen which then favors the potassium secretion, and
c) exert a direct effect on the luminal membrane resulting to increased potassium permeability (Table 1) [7].
| 1. | Increased K+/Na+-ATPase activity leading to increased potassium concentration and decreased sodium content in the cells necessary for sodium reabsorption through the ENaC. |
| 2. | Increased ENaC activity which results in increased electronegativity of the lumen leading to increased electrical gradient favoring potassium secretion. |
| 3. | Direct increased potassium permeability of luminal membrane through increased expression of ROMK channels. |
Table 1: Effects of aldosterone* on potassium homeostasis
*Aldosterone affects potassium excretion in the DCT2, CT and CCT tubules.
ROMK channel: renal outer medullary potassium channel
DCT : distal convoluted tubules
ENaC : epithelial sodium channel
CCT: cortical collecting tubules
CT: connecting tubules
Thus, the elevated serum potassium-mediated aldosterone levels along with the increased sodium and flow delivery in the so-called aldosteronesensitive potassium secretory segments are associated with increased potassium secretion through both apical ROMK and maxi-K+ channels (Figures 3 and 5a) [2,3]. The opposite mechanisms are involved in cases of reduced potassium intake (Figure 5b).
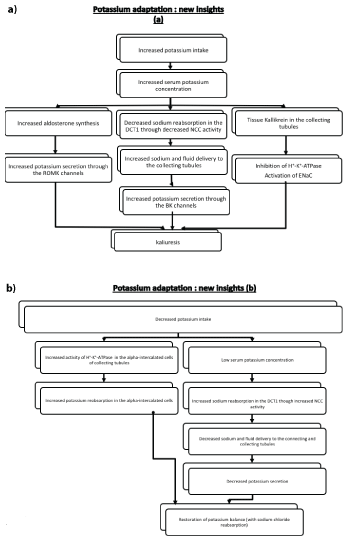
Figure 5: New insights in potassium adaptation in case of a) increased potassium intake and b) decreased potassium intake.
ROMK: renal outer medullary potassium channel
DCT1: early distal convoluted tubules
ENaC : epithethial sodium channel
NCC: Na+/Cl- -cotransporter
maxi-K+ potassium channels/BK channels: Large-conductance calciumactivated (maxi-K, BK) potassium channels
Recent studies have further delineated the underlying mechanisms of this potassium adaptation (Figures 6a and 6b). A rise in serum potassium can reduce the gradient for potassium exit via the Kir 4.1/5.1 (a unique basolateral potassium channel in the distal convoluted tubules which can be regarded as a sensor which plays a key role in the feedback control of potassium homeostasis). This channel not only recycles potassium across the cellular membrane but its activity is modulated by changes in extracellular potassium [8] resulting in increased chloride cell concentration and thus in an inhibition of the with-no-lysine (WNK) kinase and STE20 (sterile 20)/SPS-1-related proline-alanine-rich protein kinase (WNK-SPAK) cascade and finally in an inhibition of the NCC through its dephosphorylation. In this setting, it should be mentioned that the serine protease Tissue Kallikrein (TK) amplifies the excretion of a potassium load by proteolytic activation of the ENaC and inhibition of H+/K+-ATPase located in the alpha-intercalated cells (Figure 5a) [2,3] (see below for details). Interestingly this adaptation involving the sodiumchloride reabsorption in the NCC can explain at least in part the relation of potassium intake with changes in blood pressure (increased blood pressure in cases of reduced potassium intake and a reduction in blood pressure after potassium intake) [9-11].
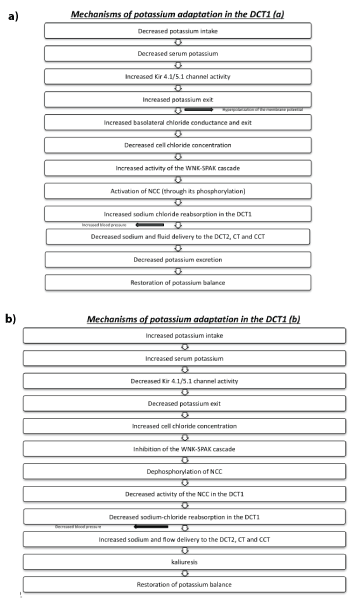
Figure 6: Mechanisms of potassium adaptation in the DCT1 in case of a) decreased potassium intake and b) increased potassium intake.
DCT1: early distal convoluted tubules
DCT2: late distal convoluted tubules
NCC: Na+/Cl- -cotransporter
CCT: cortical collecting tubules
CT: connecting tubules
WNK-SPAK: with-no-lysine kinase and STE20 (sterile 20)/SPS-1-related proline-alanine-rich protein kinase
The above mechanisms involving the cardinal role of WNK4 protein kinase 4 and its role in the NCC activity concerning potassium adaptation in the early DCT1 may also explain the so-called aldosterone paradox; thus, aldosterone can increase potassium excretion without affecting sodium reabsorption in cases of elevated potassium levels and also increase sodium reabsorption in cases of hypovolemia without affecting potassium excretion [1,12]. It was initially thought that the delivery of sodium in the distal nephron is inversely related with aldosterone concentration. Thus, in hypovolemic states, increased sodium-chloride reabsorption in the proximal tubules results in decreased sodium and flow delivery to the distal tubules and subsequently to reduced potassium secretion despite the hypovolemia-mediated increased aldosterone level. Thus, kaliuria and hypokalemia can be observed only when increased aldosterone is associated with conditions that can increase sodium and flow delivery in the distal nephron. It has recently been suggested that angiotensin II is an important modulator of this process. Thus, angiotensin II can increase NCC activity in the DCT1 through WNK and also ENaC [13-15]. Thus, in cases of hypovolemia increased angiotensin II and aldosterone lead to exaggerated sodium reabsorption. The decreased sodium and fluid delivery to the distal nephron along with the angiotensin II-mediated inhibition of ROMK channels by both WNK4-dependent and independent mechanisms are associated with decreased potassium secretion. In patients with hyperkalemia increased aldosterone levels are associated with kaliuria and restoration of potassium balance. However, the decreased angiotensin II levels (or the increased potassium levels as previously analyzed) are associated with a reduced NCC activity in the DCT1 as well as an increased activity of ROMK channels, changes that favor potassium secretion without unnecessary salt reabsorption [1,12]. Beyond aldosterone, distal potassium secretion is also depended on the rate of distal delivery of sodium and water, since:
a) increased fluid delivery is associated with increased diffusion gradient promoting additional potassium secretion (less luminal concentration of potassium). This flow-mediated increased potassium secretion is observed through the maxi-K+ potassium channels (which are quiescence in the basal state and are activated in increased flow conditions). This activation of the maxi -K+ potassium channels is mediated by changes in intracellular calcium [16-18].
b) Increased distal delivery of sodium is followed by an increased sodium reabsorption rendering the luminal potential more negative and thus promoting potassium secretion. As already mentioned, the potential (voltage) difference across the luminal membrane plays also an important role in potassium secretion, thus any condition leading to increased electronegativity of the lumen (that is reabsorption of sodium without simultaneous reabsorption of negatively non-absorbable anions, such as ketoanions, HCO- 3, ticarcillin, etc) is followed by increased potassium secretion [1]. It has been speculated that increased tubular flow is associated with increased sodium reabsorption through the ENaC, due to increased delivery of sodium but also due to mechanosensitive properties of the channel that is activated by shear stress [19]. Thus, an acute increase of serum potassium following a potassium-and proteinrich diet is associated with an increase in glomerular filtration rate (GFR) and flow rate, leading to an activation of both ENaC (through an increase in intracellular calcium) and maxi-K+ potassium channels and subsequent kaliuresis and restoration of potassium homeostasis [1].
Finally, it has recently been found that BK channels are also found in the alpha-intercalated cells of the cortical collecting tubules and can contribute to potassium secretion in cases of a positive potassium balance. Thus, a dietary potassium load can enhance BK channels expression and activity in these cells possibly due to increased WNK1 [5,18,20].
ii) As mentioned above, aldosterone-mediated mechanisms are involved in cases of reduced potassium intake (Figure 5b). In the alphaintercalated cells of the cortical collecting tubules reabsorption of potassium is observed in patients with a negative potassium balance (potassium depletion) [21]. In fact, in such cases the activity of electroneutral H+/K+- ATPase is increased leading to potassium absorption and simultaneously in H+ secretion (Figure 7). This transporter uses the energy derived from the ATP hydrolysis [22]. This transporter is implicated in the renal potassium retention during pregnancy and is progesterone-dependent [23]. Additionally, it has been found that increased adrenal progesterone production is necessary for efficient renal potassium retention in case of chronic potassium depletion independently of its role in reproduction [24].
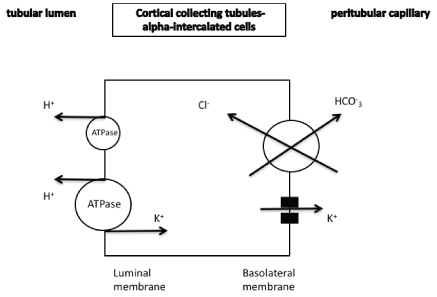
Figure 7: In the alpha-intercalated cells reabsorption of potassium is observed in cases of a negative potassium balance. Potassium secretion is also observed through the maxi-K channels.
maxi-K+ potassium channels: Large-conductance calcium-activated (maxi-K+, BK) potassium channels
It should be mentioned that reactive oxygen species are involved in apical ROMK channel inhibition in principal cells during K+ restriction. Reactive oxygen species produced by NAD(P)H oxidase stimulated by activation of the angiotensin AT1 receptor
a) inhibit protein phosphatase 2B, leading to activation (phosphorylation) of p38 and ERK, which in turn inhibit the apical ROMK channel (phosphorylation)
b) increase the protein-tyrosine kinase c-Src, which deactivates the ROMK channels (increase of tyrosine-phosphorylation) and reduce potassium excretion [25-27].
iii) Other factors also modulate renal potassium homeostasis.
It is well known that hypomagnesemia is associated with inappropriate kaliuresis and resistant hypokalemia. It has been reported that intracellular magnesium can block ROMK channels under physiological conditions and low intracellular magnesium is followed by upregulated ROMK channels and renal potassium wasting [28,29].
The renal potassium excretion is also modulated by the nature of ingested foods. Potassium-rich foods (fruits and vegetables) can increase bicarbonate concentration and intracellular pH leading to an increased ENaC activity, as well as of ROMK and maxi-K+ channels. Thus, these foods can facilitate potassium excretion [30].
Figure 8: Feed-forward control of potassium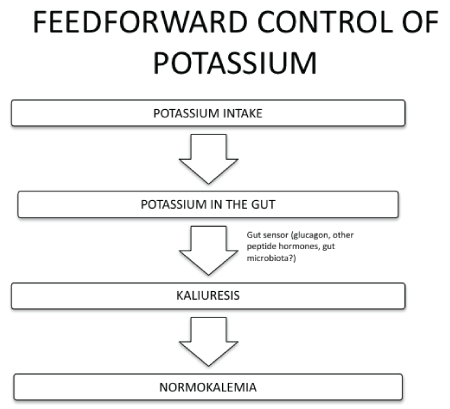
Finally, a circadian rhythm of potassium secretion has been found (potassium excretion is lower at night and in the early morning hours and is higher in the afternoon), which is possibly related to the circadian rhythm observed in gene transcripts encoding proteins affecting renal potassium homeostasis [38-40]. Studies of the physiologic circadian rhythm of renal potassium excretion provide evidence for the involvement of central nervous system and undetermined efferent kaliuretic regulatory factors other than aldosterone and plasma potassium concentration [41]. It has been found that circadian expression of H+/K+-ATPase type 2 can contribute to the circadian rhythm of urinary potassium excretion and preserve potassium homeostasis [42].
Multiple renal and non-renal mechanisms are involved in potassium homeostasis, which is a crucial ion that need to be closely controlled.
This review was conducted independently. Professor Elisaf reports personal fees from Astra-Zeneca, grants and personal fees from MSD, personal fees from Pfizer, Abbott, Sanofi-Aventis, Boehringer Ingelheim, Eli-Lilly, and GSK. The authors have given talks and attended conferences sponsored by various pharmaceutical companies, including Bristol-Myers Squibb, Pfizer, Lilly, Abbott, Amgen, AstraZeneca, Novartis, Vianex, Teva, and MSD.
For several reasons, timely diagnosis of UTIs in febrile children, including those presenting with FC is difficult. For instance, unlike adults, fever may be the sole manifestation of UTI in children, particularly during infancy. Results of this study showed that 58.8% of the patients with UTI did not have any signs or symptoms associated with UTI other than fever, demonstrating that signs and symptoms are not adequate indicators of UTI in children with FC. In an earlier study, McIntyre et al.[12] prospectively investigated the prevalence of UTI by obtaining UCs from 272 out of 307 children with FC, and compared it to a period in which UCs were not routinely obtained. Although the prevalence of UTI was substantially low (2.6%), they reported that out of children with UTI, this diagnosis was not suspected in 6. It is not surprising that in their study, the prevalence of UTI was significantly higher when compared to the period in which cultures were not routinely performed, especially in the under 2 years age group. Moreover, it has been reported that even when signs and symptoms of UTI are present, they are likely to be underrated by the physicians [18]. Thus, restricting UCs to symptomatic patients would be of low diagnostic yield, leading to missed diagnosis of UTI in a considerable number of the patients.
In conclusion, knowing the relatively high prevalence of UTI, it must be considered as a possible diagnosis for any child presenting with FC. We recommend the routine performance of UCs in this population. Future studies may focus on developing decision rules which will optimize the patient care by estimating the probability of UTI prior to further investigations.
This study was part of an MD degree dissertation conducted at Tehran University of Medical Sciences (Registration No: 22471).
Download Provisional PDF Here
Article Type: Review Article
Citation: Liontos A, Filippatos TD, Elisaf MS, Liamis G (2017) Renal Potassium Homeostasis: A Fresh Look. Int J Nephrol Kidney Failure 3(2): doi http://dx.doi.org/10.16966/2380-5498.145
Copyright: © 2017 Elisaf MS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access