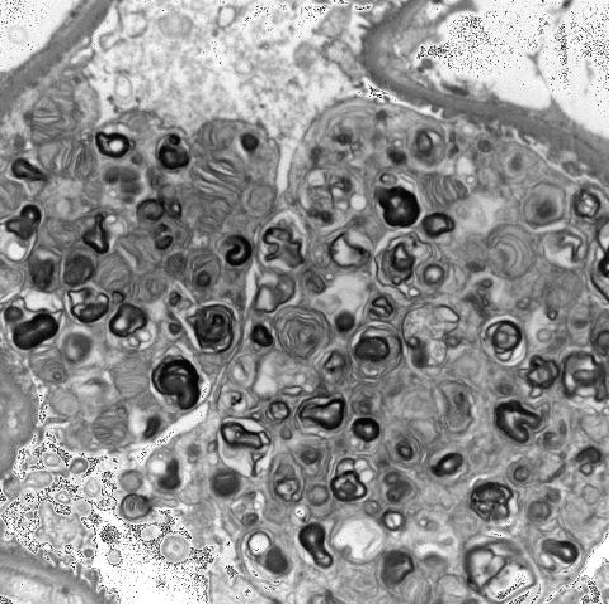
Figure 1: Electron microscopy showing extensive lamellar myelin figures in enlarged visceral epithelial cells
Shreya Goel1 Naveen K Atray2
1Lady Hardinge Medical College, New Delhi, India*Corresponding author: Shreya Goel, Lady Hardinge Medical College, New Delhi, India; E-mail: shreya_312@hotmail.com
Fabry’s disease is a rare X-linked recessive lysosomal storage disorder caused by the deficiency of an enzyme alpha galactosidase. It usually presents with paraesthesia, cutaneous lesions and death occurs due to impending cardiac or renal failure. We describe an asymptomatic 85 year old gentleman who presented to us with worsening renal function and 3+ proteinuria. His past medical history was significant for essential hypertension which was under satisfactory control. He also had history of complete heart block 20 years prior to presentation requiring a pacemaker. Hence a renal biopsy was performed which revealed findings consistent with Fabry’s disease. The family history for the same was however, negative. Over the next 3 years, his kidney function progressively deteriorated. He was started on dialysis and eventually succumbed to complications of persistent bacteremia.
Fabry’s disease results from mutations in GLA gene located on X-chromosome leading to a defect in the activity of α-Galactosidase A, a lysosomal enzyme. This causes progressive accumulation of globotriaosylceramide, a glycosphingolipid particularly in the vascular endothelium and smooth muscle cells which is usually metabolized by this enzyme.
The prevalence of Fabry disease which is estimated to range from 1:17,000 to 1:117,000 males in Caucasian population [1,2]. This X-linked recessive disorder most commonly presents as cutaneous angiokeratomas (skin rashes), acroparaesthesia (excruciating pain in hands and feet), anhidrosis (absence of sweating) and corneal dystrophy when the enzyme levels are <1%. When the enzyme activity is 1-10%, the disease runs a milder course and usually presents as renal failure and/or cardiac disease in adulthood [3]. Here we describe atypical presentation of Fabry’s disease in an elderly asymptomatic male with negative family history.
An 85 year non-smoker, non-diabetic man with well controlled essential hypertension on medication was referred to us for renal insufficiency of unknown etiology along with proteinuria in June 2011. He had history of complete heart block since 20 years which required him to be pacemaker dependent along with prior prostatectomy and an artificial urethral sphincter.
His physical exam revealed all vital signs within normal limits and a blood pressure of 120/70 mm Hg. His urinalysis was unremarkable except for 24 hour urinary protein of 1954 mg. His baseline creatinine which used to be 1.7 mg/dl had deteriorated to 2.1 mg/dl.
Renal ultrasound done in February 2011 showed echogenic kidneys consistent with chronic renal disease, no hydronephrosis and small bilateral renal calcifications. There were no cutaneous lesions or pedal edema and bilateral lungs were clear on auscultation. Ophthalmoscopy revealed macular degeneration. Serological studies including Anti-nuclear antibodies and Anti neutrophilic cytoplasmic antibody titres were negative and complement levels were within normal limits. There was no family history of kidney disease or cardiac abnormalities or any other genetic disorders. One year later, his follow up visit in September 2012 revealed worsening renal
function with serum creatinine 3.2 mg/dl ,eGFR 17ml/min/1.73 m2 for which a renal biopsy was performed in October 2012. The renal biopsy showed advanced chronicity with 65% glomerular obsolescence and moderate interstitial scarring. By light microscopy, the viable glomeruli showed distended lipid laden epithelial cells and electron microscopy showed extensive lamellar myelin figures in enlarged visceral epithelial cells (Figure 1). These changes are characteristic of Fabry’s disease.
Direct immunofluorescence suggested proteinuria and linear staining of tubular membranes with C3. Also, there was evidence of hypertensive nephropathy.
Figure 1: Electron microscopy showing extensive lamellar myelin figures in enlarged visceral epithelial cells
Measurement of alpha galactosidase level was performed. This revealed very low but detectable serum levels of α- Galactosidase A-0.03 U/L. (reference range being 0.074-0.457 U/L). Furthermore, the gene sequencing revealed one copy of c.124A>C (hemizygous). The amino acid change was reported to be p.M42L in exon 1.
This is a clinically significant missense mutation whose presence is expected to cause Fabry’s disease in a male. His family members were offered gene analysis in view of the mutation found. However, they declined to be tested. A pre-operative cardiac workup was performed which showed a normally functioning pacemaker, complete heart block and an ejection fraction of 45%. He was referred to a geneticist where he was counselled about the possibility of needing dialysis and about enzyme replacement therapy. He refused enzyme replacement therapy due to questionable benefit at this advanced stage of his chronic kidney disease and opted for peritoneal dialysis after attending dialysis modality education. We counselled him about uremic symptoms and continued to monitor his electrolytes and renal function.
In June 2013, he experienced clinical deterioration and presented with loss of sleep, energy levels and appetite. His hemoglobin had dropped, creatinine had increased to 4.9 mg/dl and eGFR had dropped to 10 ml/ min/1.73 m2 (Table 1). Since he had opted for peritoneal dialysis, a peritoneal dialysis catheter was implanted and he was initiated on peritoneal dialysis. He subsequently developed progressive thrombocytopenia and cutaneous malignancy with basal cells as well as squamous cells. There were 2 major hospital admissions in July and September 2013 for MSSA pacemaker infection.
The cardiologist opted against removing the pacemaker and he was given parenteral antibiotics. He still maintained a normal urine output. He was transitioned to in-centre hemodialysis in December 2013 due to clinical deterioration and inability to perform peritoneal dialysis at home. However he continued to have recurrent Methicillin Sensitive Staphylococcus Aureus septicemia and passed away in July 2014 after the family respected his desire to withhold any aggressive intervention and decided to stop dialysis therapy and withdraw care.
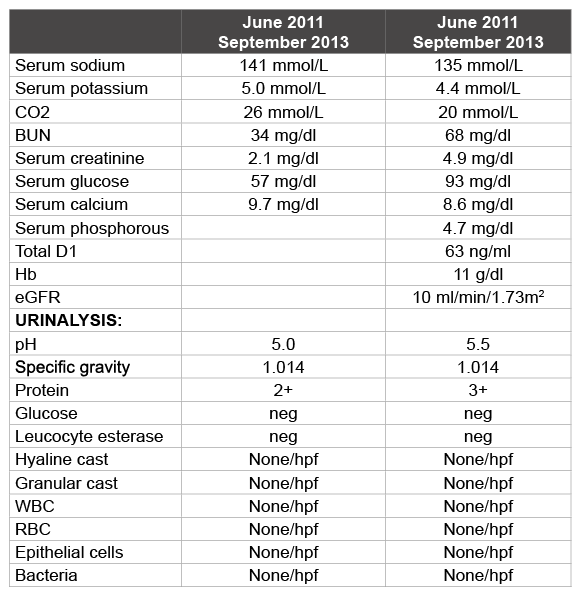
Table 1: Comparison of lab values in June 2011 at the time of presentation and September 2013
Fabry’s disease, also known as Anderson-Fabry Disease and Angiokeratoma Corporis Diffusum is a rare inherited multisystemic condition and the second most prevalent lysosomal storage disorder after Gaucher’s disease [2]. Moreover, the prevalence is probably underestimated given the non-specific manifestations of the condition due to which it is often missed or misdiagnosed. More than 370 mutations in the GLA gene (Chromosome Xq22.1) have been identified in people with Fabry’s disease. Most of these genetic changes are unique to single families [4].
Loss of function mutation that completely eliminates the enzyme activity leads to classic, severe form of the Fabry’s disease typically of childhood onset presenting with the early onset of cutaneous lesions and peripheral neuropathy. Mutations that lead to reduced enzyme activity result in milder and late onset presentation of the disease in the form of cardiac or renal failure. The case described herein had missense mutation (M42L) in exon 1. Methionine at codon 42 is highly conserved neukaryotic alpha galactosidase A orthologues. This genotype predicts a minor misfolding of alpha galactosidase A because of a small difference in hydrophobicity between methionine and leucine. This novel mutation was first described by Rosenthal et al in a 65 year old with renal variant of Fabry’s disease [5].
Fabry’s disease affects the distal tubules the most leading to decreased urinary concentrating ability and thus polyuria [6]. Polyuria and polydipsia may be among the first manifestations of Fabry renal disease [7]. It is important to note that this patient continued to maintain a good urinary output even after being started on dialysis and never required the administration of diuretics. Proteinuria which is strongly associated with renal disease progression in men and women with Fabry’s disease however, was the presenting complaint in this case [8].
The age at presentation, predominant renal presentation which was asymptomatic and atypical, absent cutaneous/neurological findings and negative family history make this case uniquely rare and atypical. In view of available treatment options, it is important for the clinicians to be aware of this disease, its typical and atypical presentations and maintain a high index of suspicion in cases presenting with unexplained proteinuria and renal insufficiency. Diagnosis of Fabry’s disease can be made from biopsy of the affected tissue, measuring the enzyme levels and by gene sequencing assay. Determination of Alpha-Gal A activity in plasma and peripheral leukocytes in males and genetic testing in females are the diagnostic goldstandards. Treatment options include enzyme replacement therapy (ERT) with Fabrazyme® (agalsidase beta). Discussion of ERT and other emerging treatments is beyond the scope of this article.
The authors declare no conflict of interest whatsoever arising out of the publication of this manuscript.
This case report did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Download Provisional PDF Here
Article Type: Case Report
Citation: Goel S, Atray NK (2016) A Curious Case of Fabry’s Disease: Delayed Renal Presentation. Int J Nephrol Kidney Failure 2(2): doi http://dx.doi. org/10.16966/2380-5498.124
Copyright: © 2016 Goel S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access