Introduction
Chemical Epitope Targeting allows for the isolation of linear and macrocyclic peptide ligands against a specific region of the protein hereby referred to as the epitope. The term epitope is borrowed from the antibody-antigen interaction lexicon [1]. When attacked by an invading foreign particle or antigen, organisms produce glycoproteins called antibodies as an immunogenic response against the antigen [2]. Antigens can be proteins such as receptors expressed on cancer cells [3], or small molecules and peptides such as hormones [4,5]. The amino acid sequence of the antigen that interacts with the antigenspecific receptor or antibody is called the epitope [6]. Epitopes can be categorized as conformational epitopes and linear epitopes. Conformational epitopes are composed of discontinuous sections of the antigen’s amino acids and interact with the antibodies based on their tertiary structures [7]. Linear epitopes, on the other hand, are formed by a continuous sequence of amino acids from the antigen and interact with antibodies based on their primary structure [8]. While a monoclonal antibody is a large, 150 kDa proteins, only a specific part of the antibody containing unique hyper variable loops called the Complementarity-Determining Region (CDR) binds to the antigen. The binding of the antigen to the antibody, its specificity, and affinity, can be dictated by non-covalent interactions like electrostatic interactions, hydrogen bonds, van der Waals forces, and hydrophobic interactions between the amino acid side chains [9]. The crystal structure of antibody with antigen shows 5-10 non-covalent interactions of the CDR of the antibody with the epitope [9]. In the Chemical Epitope Targeting strategy, we sought to find similar noncovalent interactions with the epitope, using a peptide macrocycle as an alternative for the antibody variable region. Akin to antibodies, Chemical Epitope Targeted macrocycles can reach areas inaccessible to small molecule binders of proteins, and detect subtle changes in protein structure such as phosphorylation at a single residue of a kinase [1] or a single point mutation [10].
Before the development of the Chemical Epitope Targeting strategy, some prior research was conducted to target specific areas and posttranslational modifications on proteins. Kodakek et al. [11] developed a genetic selection protocol to screen for peptides that can act as specific receptors for other peptides. They isolated a binder against the interleukin-1β (IL-1β) hormone site at which the prohormone is cut by Interleukin Converting Enzyme (ICE) 17. This was done through a lambda-reconstitution assay. Two plasmids were generated, one expressing DNA-Binding Domain (DBD) fused to the target interleukin-1β hormone peptide, and the other expressing a variable peptide library fused to a second DBD. The plasmids were co-transformed into Escherichia coli (E.Coli) cells and challenged with lambda phage. The interaction of the correct library encoded peptide with the target peptide brought together the two DBDs to reconstitute the lambda repressor and enabled lambda repressor-operator binding, which rendered the corresponding E. Coli cells immune to the infection. The peptide binders to the hormone site of IL-1β were identified from the colonies formed in presence of phage lambda. Lin et al. [12] used a different approach to generate a DNA aptamer that could select for histone H4-protein acetylated at lysine 16, implicated in regulation of gene expression, with a 60% recognition efficiency on-target. This aptamer was created to be used as a recognition ligand in singlemolecule atomic force microscope imaging of synthetic nucleosomal arrays. None of these prior methods have been as widely used as the Chemical Epitope Targeting technology.
The general method for Chemical Epitope Targeting involves screening biotin-tagged small fragments of the target protein to isolate peptide ligands for the full-length protein. The peptides developed are specific to the targeted region of the protein and have a small molecular footprint compared to a large antibody. These peptides can selectively detect phosphorylated epitopes and single amino acid point mutations, determine a species-specific sequence of malarial protein biomarker and detect a universally conserved small region of a geographically variable malarial biomarker protein [13]. In this review, we outline the general method for the Chemical Epitope Targeting screens and its applications in three areas including modulating protein function, differential detection, and protein stabilization.
Macrocyclic Peptide Libraries and Chemical Epitope Targeting Screen
For screens using the Chemical Epitope Targeting strategy, cyclic One-Bead-One-Compound (OBOC) peptide libraries are typically employed [14-16]. The library most commonly used is a heptapeptide library on TentaGel SNH2 bead (0.3 mmol/g amine loading), with the side chains (azide and alkyne) of terminal amino acids cyclized on bead by Copper (I)-catalyzed Azide-Alkyne Cycloaddition (CuAAC) reaction [13,17,18]. The library usually comprises of a five-residue variable region. The amino acids in the variable region can be any of the natural amino acids excluding cysteine and methionine. Cysteine and methionine may be oxidized during TFA cleavage and hence are not included in the library. Thus, there are 1.88 million unique sequences in this heptapeptide library. To ensure that all sequences in the library are represented, the library is made in ten-fold excess. When compared to phage display, this library has a lower diversity but can contain D-amino acids and other unnatural amino acids [13,19-21]. Incorporation of unnatural amino acids adds synthetic flexibility and increased protease stability. In addition, the free amine terminal on the cyclic peptide allows the library to be sequenced by Edman degradation without further modifications [13]. A methionine incorporated variant of this library has also been made that is cleavable by cyanogen bromide cleavage [22] and compatible with single bead sequencing by MaldiTOF/TOF [21].
Another on-bead macrocyclic peptide library that has been used is an octapeptide library, containing terminal allyl side chains that allow for on bead cyclization through RuIV-catalyzed Ring-Closing Metathesis reaction (RCM) [23, 24]. However, during the RCM reaction, excess catalyst is adsorbed in the TentaGel beads and the brown color of the beads is difficult to remove even after repeated washes with chelating reagents like sodium diethyldithiocarbamate. This makes the colorimetric detection and manual separation of the binders difficult during screens.
After the cyclization of the library, a terminal azide or alkyne is coupled to the on bead cyclic peptide for the screening (Figure 1A). The epitope of the target protein, typically a 9-30 amino acid long peptide, is synthesized with the corresponding alkyne or azide (Figure 1B). The alkyne or azide containing amino acid is either added to the C- or N-terminus of the epitope, or substitutes a natural amino acid of similar structure or hydrophobicity. The epitope is also appended with a biotin tag, separated from the peptide by a Polyethylene Glycol (PEG) linker.
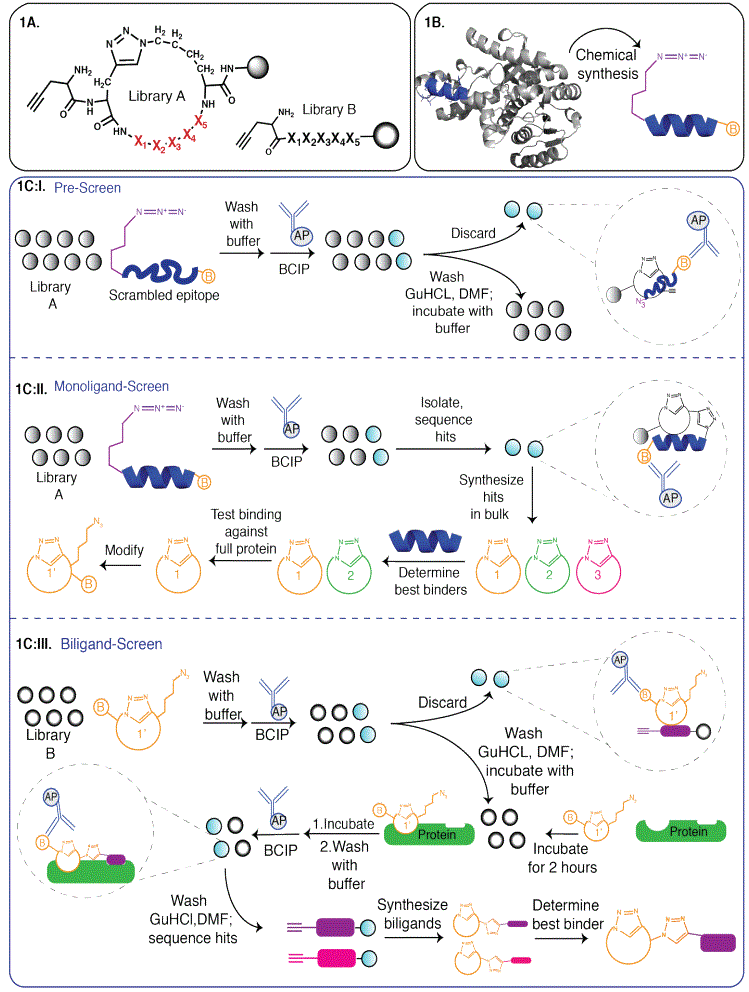
Figure 1: 1A: Macrocyclic peptide library A used in monoligand screen and linear peptide library B used in biligand screen, with Xn representing variable amino acids [13]. 1B: Target region of protein chemically synthesized as a peptide with a terminal azide and a biotin tag [13]. 1C: I. PreScreen: Cyclic library A is screened against a biotinylated scrambled version of the epitope containing an azide group. Colorimetric changes are observed in the binders after subsequent treatments with α-biotin antibody conjugated with Horseradish Peroxidase (HRP) enzyme and BCIP, a HRP substrate. The rest of the library is treated with denaturant Guanidium Hydrochloride (GuHCl) and re-equilibrated in buffer. II. MonoligandScreen: The library is screened against the biotinylated target epitope and detected using biotin antibody-HRP conjugate. The hit peptides are sequenced by Edman sequencing, re-synthesized and tested for binding to the full protein. The best peptide binder 1 is modified with a terminal azide and a biotin tag to 1’ and used to screen for a biligand. III. Biligand-Screen: 1’ is screened against library B to identify and discard non-specific binders following pre-screen procedure discussed earlier. Remaining library members are screened against the protein preincubated with 1’. The hit peptides are identified after in-situ click reaction using the biotin tag. Similar steps as the monoligand screen are followed to identify the best biligand.
There are two stages in a Chemical Epitope Targeting screen. The first stage or pre-screen involves screening the library against a scrambled version of the epitope sequence that has the amino acids shuffled in a random order. Non-specific binders are identified by treating with anti-biotin antibody conjugated to Horseradish Peroxidase (HRP) and HRP substrate 5-Bromo-4-Chloro-3-Indolyl Phosphate (BCIP) [14], which creates a turquoise colored precipitate on the bead (Figure 1C:I). The remaining library is washed with protein denaturing solutions like guanidium hydrochloride and dimethylformamide, and re-equilibrated in buffer. For the second stage or monoligand screen, the library is incubated with the epitope for several hours at room temperature. After removing non-covalently bound epitope through stringent washes under denaturing conditions, the hit beads are identified by treatment with anti-biotin antibody-HRP conjugate and subsequent BCIP treatment. After sequencing the hit binder peptides using Edman sequencing, these are synthesized without the azide or alkyne functionality (Figure 1C:II) and tested for binding to the target epitope and the full protein. Selectivity and affinity assays are done to determine the best binder [14].
A biligand or triligand (Figure 1C:III), i.e. a ligand containing two or more peptide ligands connected by a triazole linker, is developed through multiple screens of peptide libraries. The biligands and triligands have improved affinities and selectivities for the target protein due to the avidity principle, which states that combining two binders with moderate dissociation constants gives a bi- or triligand with a low dissociation constant due to cooperative interactions [25]. The shift from linear to macrocyclic peptide libraries have yielded single macrocyclic binders with good affinities without the need to further create biligands and triligands. However, ligands developed to affect enzyme function or detect subtle changes like a single residue phosphorylation, typically still need to be developed into biligands [26]. During the biligand screen, the full-length active protein is used as the target instead of the peptide epitope, so the tertiary structure of the folded protein plays a role in isolating a specific, high affinity binder. Figure 2 shows the different architectures of various peptide ligands that have been developed to date [21,27].
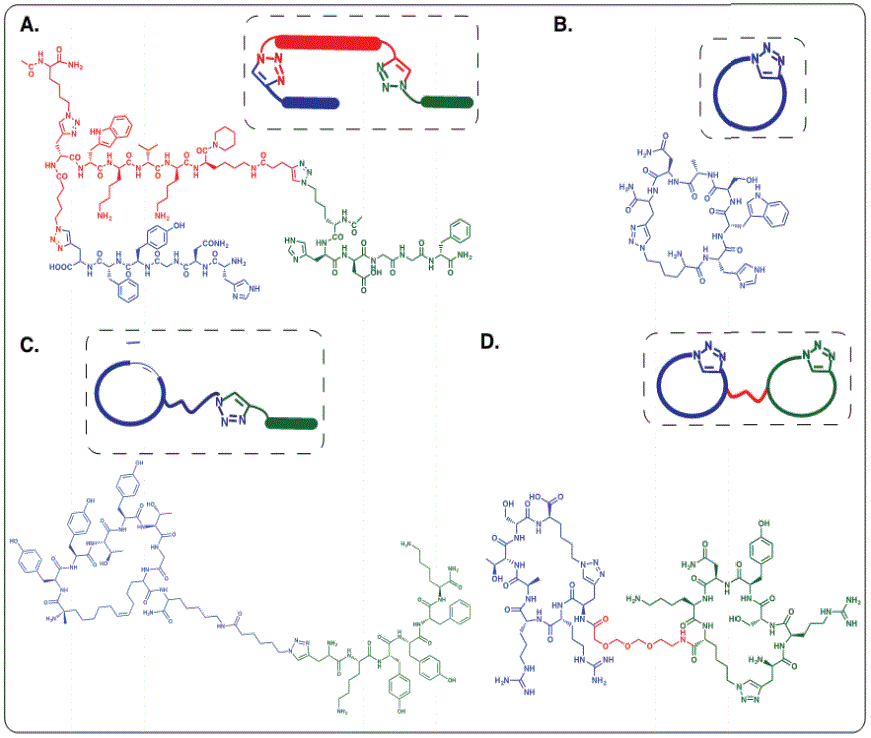
Figure 2: Architectures of different epitope targeting peptide ligands. A: Linear triligand for the C-terminal epitope of Akt2 (adjacent to pS474 residue) [1] B: Cyclic monoligand for the detection of biomarker protein Lactate Dehydrogenase for malarial virus subtype Plasmodium falciparum [13]. C: Biligand comprised of a cyclic and a linear peptide selectively binding human Akt2 [26]. D: Bicyclic ligand with optimized PEG linker for cooperative binding of discontinuous epitopes of interleukin receptor 17F [21].
Applications of Chemical Epitope Targeted Peptide Ligands
Peptide macrocyclic ligands obtained using Chemical Epitope Targeting have some of the advantages of antibodies and small molecules. The Chemical Epitope Targeted cyclic peptides do not need well-defined hydrophobic binding pockets to bind to proteins. One of the peptides developed can successfully target small, universally conserved regions of a malarial biomarker protein, whose sequence is varied in different parts of the world [28]. Peptide binders have also been isolated that can distinguish between subtle sequence differences between highly homologous protein isoforms [1,13]. All these different ligands can be improved via chemical modifications to affect potency, stability, and affinity. In this section, we shall discuss the three main applications of the Chemical Epitope Targeting that have been demonstrated in various contexts-firstly, in modulating protein function, secondly, in protein biomarker detection, and thirdly, in affecting protein folding.
Protein function modulation
Peptide ligands developed by Chemical Epitope Targeting have been used to modulate the function of oncoproteins [1,10,29] and toxins [27]. Several peptide multiligands have been developed to target different areas or isoforms of Protein Kinase B (PKB/ Akt). Akt is a serine/threonine protein kinase that is central player in the Phosphatidylinositol 3-kinase (PI3K) signaling pathway [30], who’s over expression or hyper-activation can increase the resistance of tumors to chemotherapy and radiotherapy [31]. Thus, hyperphosphorylated or E17K mutated Akt functions as an oncoprotein.
Nag et al. [1] developed peptide triligands that can both activate and inhibit Akt2 [10,29] by binding to the hydrophobic C-terminal end, where the Ser474 residue is located. Ser 474 phosphorylation allosterically activates the protein and enhances the kinase activity 10- fold [32,33]. For constructing the epitope, a peptide consisting amino acids 450-481 of Akt2 including phosphorylated Ser474 (pS474) was first synthesized. Then, a small molecule containing a zincdipicolylamine complex, an azide and a biotin label was synthesized. Co-ordinate covalent interaction of the binuclear zinc complex to the phosphate group on pS474 created a modified peptide with an azide that was used as the epitope in the screen. The epitope was screened against an alkyne containing D-amino acid hexapeptide linear library to obtain a primary ligand (KD 3µM). The final two triligands, N-tri and C-tri, developed through subsequent screens, had higher affinity and high selectivity for Akt2. The triligand C-tri demonstrated a 10:1 selectivity for Akt2 over the 85% homologous Akt1. Surface Plasmon Resonance experiments were performed to demonstrate the high binding affinity of N-tri for Akt2 (KD 20 nM). Interestingly, the two ligands had reverse effects on the Akt kinase activity, with C-tri inhibiting Akt2 (EC50 4 µM), and N-tri increasing the Akt2 kinase activity.
Henning et al. [29] further developed N-tri and C-tri [1] into proteolysis targeting chimeric molecules (PROTACs) to promote rapid degradation of Akt in cancer cells. N-tri was conjugated with a cell penetrating TAT sequence [34] and a peptide ligand that binds to the E3 ubiquitin ligase Von Hippel Lindau protein (VHL) from the Hypoxia-Inducible Factor (HIF-1) protein [35], and induced Akt2 degradation. Treatment of OVCAR3 cells with this peptide construct showed dose-dependent decrease in Akt2 levels with an EC50 of 128 µM [29,36].
Chemical Epitope Targeting technology was generalized by Das et al. [13] to eliminate the need for the epitope to have a specific functionality like a phosphate. This was achieved by substituting certain amino acids like lysine and arginine with an unnatural amino acid bearing an azide group in the peptide epitope. For the epitope to present an alkyne instead, amino acids isoleucine and valine can be replaced with an alkyne bearing unnatural amino acid [10,27]. For developing binders against single point mutations, the substitution of the unnatural amino acids bearing the azide or alkyne group is positioned 3 to 4 residues away from the mutated residue. This generalized technique was employed [13] to develop an inhibitor against E17K mutated Akt1 that exhibited a 10:1 selectivity for the mutant protein over the Wild-Type (WT) protein. E17K Akt1 is an oncogenic variant of Akt1 [37], as this mutation in the Pleckstrin Homology (PH) domain of Akt1 increases mutant Akt1’s affinity for phospholipid Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and facilitates membrane binding, thereby activating the PI3K-Akt pathway. Deyle et al. [10] synthesized a 33 amino acid long peptide epitope of Akt1 containing the E17K mutation, a biotin tag and substituted an isoleucine with propargylglycine for in situ click screen. The epitope was screened against an azide containing D-peptide library with 1.88 million unique sequences. The hits demonstrated a high affinity for E17K Akt1 (KD 54nM) as opposed to WT Akt1 (KD 1.2µM). Ligand-directed labeling experiments confirmed that the ligand bound to the targeted region of the protein. Multicolor fluorescence microscopy experiments were performed to demonstrate the selective colocalization of the Cy5 labeled ligand with GFP tagged E17K PH domain inHEK-293T cells. While the original ligand could not disrupt the strong interaction between the PH domain of E17K Akt1 and PIP3 , the triligand, developed through multiple screens against Akt1 PH domain as target, had enough steric hindrance to inhibit the interaction.
Taking advantage of the dynamic nature of Botulinum Neurotoxin (BoNT) and its entry mechanism, Farrow et al. [27] developed a competitive inhibitor for BoNT serotype A targeted at its occluded active site. BoNT is a chemodenervating zinc-dependent protease that intoxicates the cell by selectively binding to neural receptors, entering the cell through receptor-mediated endocytosis, escaping the endosome using pH-induced translocation, and cleaving its SNAP-25 substrate in the cytosol [38]. The protein contains a receptor-binding heavy chain which is disulfide-linked to a catalytic Light Chain (LC). This disulfide bond needs to be intact for the toxin to enter the cell but the LC is structurally occluded at that stage. In the cytosol, the disulfide link is reduced and the LC can catalytically cleave the SNAP25 substrate [39-41], exposing and making the active site druggable only inside the cell [42-44]. This biological mechanism was harnessed by Farrow et al. [27] to develop a substrate-mimicking bicyclic peptide inhibitor with an IC50 of 165 pM [27]. The first cycle in the ligand, Inh1, was a helical substrate mimetic of the SNAP-25 substrate [45] that can bind near the Botox active site once the ligand enters the cell. A second cyclic ligand was developed against surface exposed BoNT LC residues 166-179 that would mediate the entry of the ligand into the cell. An in situ click screen was performed to reveal the correct length and nature of the linker connecting the two cyclic ligands so that the bicyclic ligand bound with high affinity to the BoNT. The bicyclic ligand was modified with a spontaneously translocating peptide sequence for BoNT independent penetration of the cells [46]. The final ligand demonstrated significant protective effects to BoNT intoxication of neurons at low nanomolar concentrations [27].
Differential detection of proteins
In addition to affecting protein function, Chemical Epitope Targeting has been used for differential detection such as distinguishing between receptor isoforms [21] or homologous proteins of different malarial species [13]. This technique has been applied to identify ligands that can detect malarial biomarkers found in infected human blood, namely Lactate Dehydrogenase (LDH) and Histidine Rich Protein 2 (HRP2). By screening against an epitope that was distinct in LDH of different malarial species, Plasmodium falciparum (Pf) and Plasmodium vivax (Pv), Das et al. [13] developed peptide ligands that could distinguish between Pf LDH and Pv LDH. A binder with a 13:1 selectivity for Pf LDH over Pv LDH was isolated that did not show any cross-reactive binding to human LDH.
Furthermore, Das et al. [13] screened for a binder selective for a universally conserved motif of the PfHRP2 protein [13]. PfHRP2 protein is an intrinsically disordered protein whose sequence is varied in different geographical regions of the world [28,47]. It consists of repetitive motifs containing numerous histidine and alanine residues. Two conserved motifs near the C-terminus were used as a target epitope which was screened against a cyclic peptide library. A binder with a strong affinity (KD 54.3 nM) and selectivity for PfHRP2 was identified [13].
Polypeptide macrocycles have been developed for the differential detection of the cytokines interleukin-17A (IL-17A) and interleukin17F (IL-17F) [21]. These two pro-inflammatory cytokines, sharing a sequence homology of about 55% [48], are secreted by immune cells and are associated with multiple immune and autoimmune diseases [49,50]. Lai et al. [21] synthesized two discontinuous epitopes for IL-17F and one continuous epitope for IL-17A and screened for binders. They isolated a binder with a 3:1 selectivity for IL-17A over IL-17F. The two binders for the two IL-17F epitopes were linked with a Polyethylene Glycol (PEG) linker to give a cooperative bicyclic biligand that bound to the discontinuous epitopes. Fluorescence polarization experiments demonstrated the biligand (KD 252 pM) had an improved affinity (17 fold higher) and specificity (2.5 fold higher) compared to the individual monoligands [21].
Protein folding
Another application of the Chemical Epitope Targeting technology has been to generate a cyclic peptide that influences protein folding in Superoxide Dismutase 1 (SOD1). Stress factors in the cell produce free radicals, and the highly reactive oxygen radical is converted by SOD1 into hydrogen peroxide and oxygen [51]. About 160 point mutations are known to occur in SOD that lower its stability and promotes misfolding [52], leading to neurological diseases like Amyotrophic Lateral Sclerosis (ALS) [52-55]. Bunck et al. [51] targeted an electrostatic loop of SOD (residues 121-141) involved in destabilization of SOD1 structure [56] and identified a ligand (EC50 8 µM) that bound to the loop. Ligand binding to the electrostatic loop resulted in a tightening of the overall structure and stabilized the folded WT apoprotein. Interestingly, incubating the ligand with the apoforms of mutant G85R, G93A or D90A SOD1 also led to stabilization of the folded protein structures, as indicated by the lowering of the hydrodynamic radii in Dynamic Light Scattering experiments. The peptide ligand therefore acts as a chemical chaperone for folding of both WT and mutant SOD [51,57].
Conclusion
Initially developed with the aim of targeting a peptide epitope adjacent to phosphate functionality, Chemical Epitope Targeting has now evolved into a universally applicable technique. It has been successful in detecting very specific regions of a protein, a goal rarely achieved by any other library screening process of peptides, small molecules and nucleic acid aptamers. Peptide binders have been developed for a host of proteins, including proteins belonging to families as varied as intracellular kinases (Akt1, Akt1 PH domain, phosphorylated Akt2) [1,10], essential enzymes (SOD) [51], secreted blood proteins (PfLDH, PvLDH, PfHRP2) [13], toxins (BoNT) [27], viral envelope proteins (L1R) [13] and cell surface receptors (IL17A and IL17F) [21]. Chemical Targeting Technology has been utilized for sophisticated applications, such as increasing or decreasing Akt2 kinase activity, rescuing neurons from BoNT toxin effect, and stabilizing protein folding state for apoforms for mutant SOD.
An important development with wide implications has been the success in applying this technology in tandem to the PROTAC technology [29]. When used together, these can allow for selective degradation of a mutated protein by isolating a peptide binder for the protein and making it into a PROTAC. Another important development has been the successful targeting of discontinuous epitopes [21,27], which will probably play a major role in further applications of this technology
Acknowledgement
Dedicated to Professor James R. Heath, President, Institute for Systems Biology
