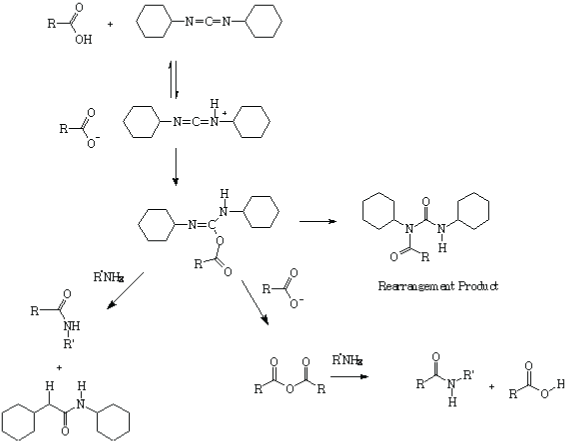
Scheme 1: Mechanism of amidation using DCC as a coupling reagent, and the formation of the rearrangement product.

Ezequias P Siqueira1* Isabela P Ceravolo2 Markus Kohlhoff1 Antoniana U Krettli2 Carlos L Zani1
1Laboratório de Química de Produtos Naturais Bioativos, Instituto René Rachou, Fundação Oswaldo Cruz, Belo Horizonte Minas Gerais, Brazil*Corresponding author: Ezequias P Siqueira, Laboratório de Química de Produtos Naturais Bioativos, Instituto René Rachou, Fundação Oswaldo Cruz, Av. Augusto de lima 1715, Barro Preto, 30190002, Belo Horizonte Minas Gerais, Brazil, Tel: +55 31 33497845; E-mail: ezequias@minas.fiocruz.br
Malaria, a disease caused by the Plasmodium parasite, has spread globally, mainly in the African subcontinent. Species of parasites resistant to usual drugs like chloroquine, used to treat malaria, have led research groups to search for new drugs to expand the current therapeutic arsenal. Fourteen 2-methyl-3-carboxylnaphtho[2,3-b]furan quinone derivatives (FNQ) were synthesized and evaluated using cultures of Plasmodium falciparum in vitro. From all FNQ derivatives tested, six of them (4,7-10,14) showed IC50 values ranging from 0.05 µM to 11.3 µM. The glutamineFNQ (9) and spermidine-FNQ derivatives (14) were the best antiplasmodial molecules tested, with selectivity indexes of 56 and 75, respectively. Our data suggests these two compounds are appropriate candidates for investigation as potential antimalarial drugs against drug-resistant Plasmodium species.
Naphthofuran quinones; Plasmodium falciparum; Malaria; Drug discovery
Malaria is one of the main causes of death worldwide. According to the World Malaria Report 2017, a publication of the World Health Organization, 91 countries reported a total of 216 million cases of malaria and 445,000 deaths in 2016. Malaria afflicts tropical and subtropical countries around the world, but is particularly prevalent on the African continent, where most cases are reported [1]. Between 2000-2015, malaria incidence rates fell by 37% globally, and by 42% in Africa. During this same period, malaria mortality rates fell by 60% globally and by 66% Africa [2]. Several factors, including increased research financial support, effective vector control, effective malaria diagnosis and immediate and effective treatment regimens have been important factors in decreasing the burden of malaria-affected countries. The appearance and spread of resistant parasites to multiple antimalarial agents represent one of the greatest challenges to controlling the disease. Malarial drug resistance is associated with increased morbidity, mortality, and poverty in many countries where malaria is endemic [3,4]. Thus, it imposes a constant pressure to continue finding new antimalarial drugs for treating the several forms of malaria [5].
Strategies are being developed to rationalize anti-malarial drug design, especially by disrupting essential parasite biosynthetic pathways, e.g., inhibition of dihydroorotate dehydrogenase [6], Plasmodium falciparum lactate/proton symporter [7], parasite proteases [8], triose phosphate isomerase, glyceraldehyde-3-phosphate dehydrogenase, and aldolase [9]. Hundreds of quinone derivatives have been investigated for their antimalarial activity [10], with atovaquone reaching clinical use, especially in combination with proguanil, which is sold under the trade name Malarone®. Our research group has worked in the search of new drug leads for malaria and others tropical diseases. Herein, we present the synthesis and in vitro anti-plasmodial activity testing results of fourteen 2-methyl-3-carboxyl-naphtho[2,3-b]furan quinone derivatives (FNQ).
General: All solvents were P.A. grade, previously purified and maintained over molecular sieves 4 Å. Reactions were conducted under anhydrous conditions and N2 atmosphere in flame dried glassware if appropriate. Nuclear Magnetic Resonance (NMR) spectra 1H, 13C were measured on BRUKER Advance Instrument, at 400 and 100 MHz, respectively at 27°C with deuterated chloroform or deuterated d6-dimethyl sulfoxide (DMSO) as solvents, and tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are given in ppm and coupling constant (J) in Hertz. Infra-red (IR) was recorded using a Shimadzu FTIR-8400 and KBr pellets and the data expressed in cm-1. High Resolution Mass Spectrometry (HRMS) were measured on maXis ETD high-resolution ESI-QTOF mass spectrometer (Bruker) controlled by the Compass 1.5 software package (Bruker). Data dependent fragment spectra were recorded using a collision energy range between 15 and 60 eV. Ion cooler settings were optimized within a 40-1000 m/z range using a calibrant solution of 1 mM sodium formate in 50% 2-propanol. HRMS data showed as adduct of M+H and/or M+Na and the deviation of the theoretical molecular weight in ppm. Purifications of the compounds were performed using preparative Reverse Phase High Performance Liquid Chromatography (RP-HPLC), using a preparative Shim-pack C18 column and Shimadzu HPLC System and CH3CN-H2 O/ MeOH-H2O as solvents or normal phase (silica gel 60) using hexane/ethyl acetate as solvents. Reactions were monitored by TLC using HF254 silica plates (Merck) and MeOH/ dichloromethane (DCM) as eluents and spots visualized under visible light or in a UV chamber. Lipophilicity (log P) and solubility in water (log S) were predicted theoretically by means of DataWarrior Software V.4.7.2 (www.openmolecules.org/ propertyexplorer/).
Synthesis: Compound 1 was prepared as previously described by Hu et al. [11]. Compound 2: In a 100 mL capacity flask were placed 2.84 g of 1 (10 mmol) and 100 mL of glacial acetic acid, 1 ml of water and 0.1 mL of conc. HCl. The system was put under reflux for 3 hours and then cooled on ice. The precipitated yellow solid was filtered through sintered filter, collected and dried to furnish 2.50 g of 2 (9.8 mmol). Compound 3: In a dry flask 100 mL were placed 256 mg of 2 (1 mmol), 412 mg of DCC (N,N’-Dicyclohexylcarbodiimide, 2 mmol) and 30 mL of anhydrous N,N-dimethylformamide (DMF). The mixture was stirred with a magnetic bar for 4 hours at room temperature until its color changed to dark yellow. The mixture was dried under vacuum and the residue was purified in preparative silica gel column (I.D 3.0 cm; length 45 cm) using hexane-ethyl acetate gradient. Fractions of 50 mL were collected and analyzed by HRMS. After dried over vacuum it yielded 203 mg of 3 (0.44 mmol). Compounds 4-13: To sealed dried vials was added 1.0 g of Wang resin (0.6-1.0 mmol/g, Sigma-Aldrich), 520 mg PyBOP (1.0 mmol) and 0.6-0.8 mmol of the respective FMOC (Fluorenylmethyloxycarbonyl)-protected aminoacids, HOOCaa-FMOC (Sigma-Aldrich), in 30 mL of anhydrous N-methyl morpholine solution (NMM) in DMF (3%). The system was stirred for 4-48 hours. After filtration of the reaction mixture, the resin was washed three times with anhydrous DMF, and stirred with a solution of piperidine in DMF (20%) for 10-60 minutes. The resin was again filtered and washed as above, to yield the Wang resin loaded with the FMOC protected aminoacid. Each of these Wang-aa was added to sealed flasks containing 25.6 mg (0.1 mmol) of 2 and 570 mg PyBOP (1.1 mmol) in 5 mL of anhydrous solution of N-methyl morpholine solution (NMM) at 3% in DMF. This system was stirred for 4-16 hours, filtered and washed with DMF. The FNQ-aa-Wang resin was washed with TFA:DCM 1:1 during 1-3 hours to cleave the FNQ-aa and the yellow acidic liquid collected. After elimination of the solvent and TFA the crude product was purified by RPHPLC. Chromatographic separations were performed in C18 Shim-pack preparative column (I.D. 2.5 cm; length 25 cm) and H2O/CH3 CN 10-100% at 1 mL/min in 50 minutes, yielding the respective pure FNQ-aa (Table 1). Compound 14: In a dry sealed vial, 1100 mg of PyBOP (Benzotriazol-1-yloxytripyrrolidino-phosphonium, 2.1 mmol) were mixed with 570 mg of 2 (~2.2 mmol) and a 3% solution of anhydrous N-methyl morpholine (NMM) in DMF (50 mL) added. After complete dissolution, 200 µL (72.3 mg, ~2.0 mmol) of spermidine were added and stirred for 30 minutes at room temperature. A brown solid was formed. The mixture was filtered, and the liquid phase was evaporated to dryness and purified by RP-HPLC using C18 Shimpack column and H2O/CH3 CN 10-100% in 50 minutes yielding ~200 mg of 14 (0.32 mmol).
 |
|||||||
| NFQ Number | Molecular Structure | Yield (%) | LogP+ | LogS++ | IC50 (µM)* | MDL50 (µM)** | SI*** |
| 1 | 26 | 2.9393 | -5.221 | 1.4 ± 0.7 | 12.0 ± 6.0 | 8 | |
| 2 | 98 | 2.1051 | -4.793 | >39.0 | NT | NA | |
| 3 | 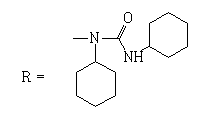 |
44 | 5.2224 | -8.032 | 2.8 ± 0.7 | 14.0 ± 4.3 | 5 |
| 4 | 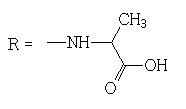 |
36 | 1.5481 | -4.938 | 11.3 ± 2.8 | 174.2 ± 38.8 | 15 |
| 5 | 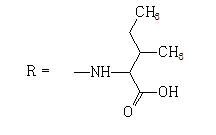 |
24 | 2.6752 | -5.638 | 6.0 ± 1.6 | 53.6 ± 12.2 | 9 |
| 6 | 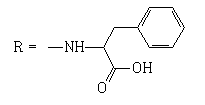 |
35 | 2.99 | -6.073 | >25.0 | NT | NA |
| 7 | 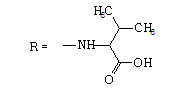 |
15 | 2.2208 | -5.368 | 9.6 ± 0.6 | 157.9 ± 46.2 | 16 |
| 8 | 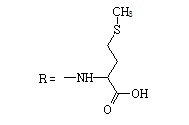 |
9 | 2.1517 | -5.583 | 7.7 ± 4.6 | 96.5 ± 11.6 | 12 |
| 9 | 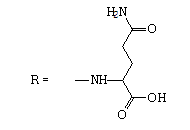 |
12 | 0.7828 | -5.029 | 2.0 ± 0.2 | 111.9 ± 11.0 | 56 |
| 10 | 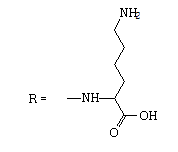 |
26 | -0.14 | -5.317 | 0.7 ± 0.1 | 19.5 ± 4.4 | 28 |
| 11 | 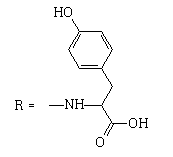 |
19 | 2.6443 | -5.777 | >23.8 | NT | NA |
| 12 | 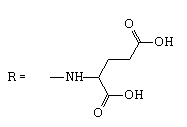 |
11 | 1.1806 | -4.953 | 9.3 ± 3.3 | 57.1 ± 14.8 | 6 |
| 13 | 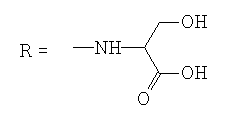 |
18 | 0.6214 | -4.431 | 4.4 ± 0.1 | 26.2 ± 4.9 | 6 |
| 14 | 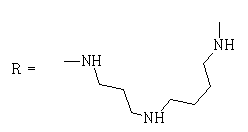 |
32 | 4.7495 | -9.781 | 0.05 ± 0.02 | 3.8 ± 1.0 | 75 |
| Chloroquine | - | - | - | - | 0.3 ± 0.1 | 1428.7 ± 68.7 | 4271 |
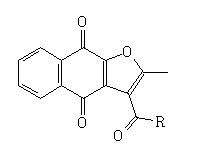
Table 1: Yield, theoretical predicted physicochemical properties, In vitro activity, cytotoxicity and selectivity index of the FNQ tested against blood
forms of Plasmodium falciparum resistant clone
In vitro results are representative of at least two independent experiments.
+ Theoretical predicted lipophilicity described as log Poctanol/water where P =([solute]octanol/[solute]water).
++ Theoretical solubility in water described as log S where S=solubility in mol/l, pH=7.5, 25°C
*Antiplasmodial activity evaluated using SYBR test.
**Cytotoxicity evaluated by the incorporation of neutral red uptake assay.
***Selectivity Index obtained from the ratio MDL50 and IC50, where SI ≤ 10 was indicative of toxicity.
NT: Not tested; NA: not applicable.
Biological tests
In vitro assay with Plasmodium falciparum culture: The compounds were tested against the P. falciparum parasite erytrocytic asexual stages using a chloroquine-resistant and mefloquine-sensitive W2 clone [12] cultured at 37°C as previously described [13]. The activity was measured using the SYBR assay with the parasite suspension (0.5% parasitemia and 2% hematocrit), as previously described [14]. Briefly, the test compounds, in serial dilutions, were incubated in “U” bottom 96-wells plates. After 48 h at 37°C, the culture supernatant was removed and replaced by 100 µL of lysis buffer solution [Tris (20 mM; pH 7.5), EDTA (5 mM), saponin (0.008%; wt/vol), and Triton X-100 (0.08%; vol/vol)] followed by addition of 0.2 µL/ mL SYBR Safe (Sigma-Aldrich, Carlsbad, CA, USA). The plate content was transferred to a flat bottom plate and incubated in the dark for 30 min. The plate was read in a fluorometer (Synergy H4 Hibrid Reader, Biotek) with excitation at 485 nm and emission of 535 mm. In all tests, the compounds activities were expressed by the 50% inhibitory concentration of the parasite growth (IC50) when compared to the drugfree controls and estimated using the curve-fitting software Origin 8.0 (OriginLab Corporation, Northampton, MA, USA). Compounds with IC50 great than 20 µM were classified as not active, and less than 20 µM were considered as active. The chloroquine was used as antimalarial reference drug.
Toxicity to a mammalian cell line: The monkey kidney cell line (BGM) (ATCC, Manassas, VA, USA) was used forthe cytotoxicity assays, and maintained as suggested by the manufacturers. The cells were cultured in 75 cm2 bottles with RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum and 40 mg/L gentamicin in a 5% CO2 atmosphere at 37°C. For the in vitro tests, a confluent cell monolayer was treated with trypsin and the cells distributed in a flat-bottomed 96-well plate (2 × 105 cells/mL) and incubated for 18 h at 37°C to ensure cell adherence. The BGM cells were incubated with 20 µL of the drugs at different concentrations (≤ 1,000 µg/mL) for 24 h in a 5% CO2 at 37°C. The cell viability was expressed as the percentage of control absorbance obtained from the untreated cells after subtracting the appropriate background. The drug lethal dose of cells was determined using at least duplicate tests to calculate the dose that killed 50% of the cells (MLD50), as described by do Céu de Madureira et al. [15].
The neutral red uptake assay was used to evaluate the lysosomal integrity and distinguishes live cells from dead by its ability of incorporate the dye [16]. Briefly, to each well was added 0.2 mL medium containing 50 µg/mL of neutral red solution. The plate was incubated for another 3 h at 37°C to allow for the uptake of the vital dye into the lysosomes of viable uninjured cells. After removal of medium, 200 µL of a mixture of 0.5% formaldehyde-1% CaCl2 was added to the cells, and incubated by 5 min. The supernatant was removed and 100 µL of a solution of 1% acetic acid-50% ethanol was added to each well to extract the dye. After homogenization, the optical density of each well of the plate was measured using a 540 nm wavelength on a spectrophotometer. This absorbance has shown a linear relationship with the number of surviving cells. The ratio between drug cytotoxicity (MLD50 BGM) and activity (IC50 W2) was used to estimate the selective index (SI), as shown before [17], where SI less than 10 was indicative of toxicity.
Our studies aimed at synthesizing and testing the in vitro activity of FNQ derivatives against P. falciparum. According to Basselin et al. and Carrillo et al. [18,19], amino acids, amine derivatives, and endogenous polyamines, such as spermidine and putrescine play important roles as molecular transporters and in maintaining cell homeostasis in the different life-cycle stages of different protozoan parasites. Considering that some quinones possess potent antimalarial activity, we speculated that the conjugation of an FNQ with an amino acid/spermidine could help to increase its intracellular concentration, resulting in more potent or selective compounds. We thus envisaged the condensation of the amino group of several amino acids with the carboxyl group in FNQ (2). Subsequently, starting from the easily prepared ester 1, using an established protocol the corresponding acid (2) was obtained after acid hydrolysis. This FNQ carries a carboxyl group and was used as a building block to produce corresponding amides with ten different amino acids.
Previous experiments were performed using DCC as the coupling reagent; however, we observed after purification and analysis of the spectroscopic data the occurrence of a rearrangement to afford compound 3. The rearrangement of the acidic-DCC specie halts to complete the amidation reaction [20] (Scheme 1).
Scheme 1: Mechanism of amidation using DCC as a coupling reagent, and the formation of the rearrangement product.
We adopted solid-phase peptide synthesis using FMOCprotected methodology for construction of ten FNQ-amino acid derivatives [21]. Briefly, Wang resins preloaded with different N-FMOC-protected amino acids (aa) were treated with piperidine to remove the FMOC protecting group. The unprotected amino group was condensed with FNQ with the aid of PyBOP in the presence of N-methyl morpholine. Finally, the condensation products were released from the resin with TFA (Scheme 2) and purified by reversed phase chromatography to yield the pure compounds 4-13.
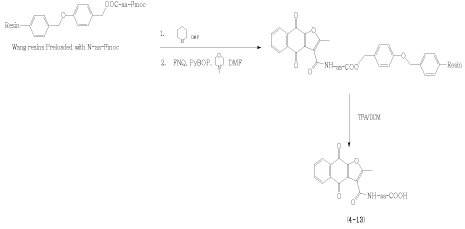
Scheme 2: The synthetic route of the compounds 4-13 (amino acid derivatives - aa) using PyBOP as a coupling reagent in solid phase synthesis.
As a result of a large screening of synthetic compounds against P. falciparum conducted by several groups worldwide in 2010, it was found that the amide obtained from FNQ (2) and isopentylamine, despite its relatively low activity (IC50 of 1.2 µM against the Pf3D7a clone), showed a synergistic effect with artemisinin, indicating a dramatic reduction of its IC50 (22 nM) in the presence of this antimalarial at its IC10 concentration (20 nM) [22]. These results led us to evaluate the activity of the compound prepared by condensing FNQ with spermidine. Analysis of the spectroscopic data of the synthetic products showed that they corresponded to a mixture of mono- (minority) and di- (majority) FNQ-spermidine derivatives, such as compound 14.
The yields for all compounds synthesized, and the antimalarial activity data are summarized in table 1. According to table 1, low yields of the FNQ-amino acid derivatives (4-13) were observed, varying from 9 to 36%. The global reaction occurred in several steps starting from a small amount of material (0.1 mmol). The experimental conditions were not optimized to improve yield, and it was noticed that starting material, as well as some byproducts and a final complex crude product were purified. All these non-optimized steps contributed to a low yield.
The results obtained clearly show the structure-activity dependence of the ligands to the core of the FNQ. Compounds where IC50 was greater than 20 µM were classified as inactive and their respective MDL50 and SI values were not determined (2, 6 and 11), because the selectivity index is only determined for active samples. The active compounds, where IC50 values were less than 20 µM showed SI values ranging from 5 to 75.
Several factors inherent to the structure of the drug contribute to biological activity. As biological assays occur in aqueous medium, the availability of the drug depends on their solubility in aqueous medium and their respective lipophilicity. Very hydrophobic compounds (log P>5) are difficult to dissolve in aqueous medium and have low assimilation because of their inherent low capacity to travel across membranes of the parasite to act inside the cytoplasm and/or nucleus [23,24]. In the same way, the stereochemistry of the substance is a crucial condition to allow recognition by enzymes.
When testing the FNQ-amino acids series we noticed that those containing aryl residues were not active (6, 11); alkyl residues showed dependence on their structure. For example, the SI of isopropyl (7) is greater than SI of methyl (4) is greater than SI of sec-butyl (5). Amine terminal residues (9, 10) showed SI values higher than the acidic terminal (12). Hydroxy terminal (13) residues were shown to be toxic.
Out of the amino acids series, the compounds 1¸2, 3 and 14 represent a myriad of functional groups and activities ranging from 50 nM and SI values of 75 (14) or were toxic (2). In terms of hydrophobicity, the compounds where SI>10 (4, 7-10, 14) showed a large range of values for log P, varying from -0.14 to 4.75 (10 and 14, respectively); this means that an order of hydrophobicity of almost 105 times lies between them. In terms of solubility in the aqueous medium, the range for log S was from -4.94 to -9.78 (4, 14 respectively) and an order from 11.3 µM to 0.14 nM.
Our preliminary studies for drug discovery against neglected diseases and/or diseases where the therapeutic arsenal is not supported led us to present the results of the activity from FNQ derivatives as candidates for drug development. It was shown that modifications in the lateral chain of the FNQ core modified the activity results; thus, this position on the FNQ core is a crucial site in the mechanism of action of the drug against parasites.
We determined that the glutamine-FNQ (9) and the spermidine-FNQ derivatives (14) were good candidates for study, because their SI levels were higher than 10. These drugs could be used as co-adjuvants to increase the potency of reference drugs against resistant parasites, but studies regarding their mechanism of action need to be performed.
ethyl 2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b] furan-3-carboxylate
1 H NMR (400 MHz, chloroform-d) δ ppm: 1.45 (t, J=7.20 Hz, 3 H); 2.72 (s, 3 H); 4.44 (q, J=7.17 Hz, 2 H); 7.71-7.79 (m, 2 H); 8.14-8.22 (m, 2 H).
13CNMR (101 MHz, chloroform-d) δ ppm: 14.33; 14.36; 61.68; 113.93; 126.65; 127.56; 128.38; 131.67; 133.80; 133.88; 134.25; 151.49; 162.17; 164.54; 173.63; 178.81.
HRMS–C16H12O5 (M+H)=285.0763 (1.9 ppm); (M+Na)=307.0582 (2.5 ppm).
IR–KBr (cm-1): 3000, 2973, 2943, 1709, 1699, 1683, 1654, 990, 717.
2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-3- carboxylic acid
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.67 (s, 3 H); 7.81 - 7.91 (m, 2 H); 8.08 - 8.10 (m, 2 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 13.65; 113.62; 126.10; 126.79; 127.39; 131.36; 132.98; 134.30; 134.34; 150.88; 162.58; 163.88; 172.72; 179.51.
HRMS–C14H8O5 (M+H)=257.0449 (1.8 ppm); (M+Na)=279.0267 (1.0ppm).
IR–KBr (cm-1): 3488, 3133, 1742, 1674, 1653, 996, 724.
(N-(cyclohexylcarbamoyl)-N-cyclohexyl-2-methyl-4,9- dioxo-4,9-dihydronaphtho[2,3-b] furan-3-carboxamide)
1H NMR (400 MHz, chloroform-d) δ ppm: 0.62-1.22 (m, 5H); 1.24-1.50 (m, 5H); 1.57-1.75 (m, 4H); 1.79-1.93 (m, 4H); 1-99-2.01 (m, 2 H); 2.50 (s, 3 H); 3.42 - 3.49 (m, 1 H); 4.24 (m, 1H); 6.18 (br, s 1H); 7.74 - 7.83 (m, 2 H); 8.09 - 8.15 (m, 1 H); 8.20 - 8.25 (m, 1 H).
13C NMR (101 MHz, chloroform-d) δ ppm: 12.81; 24.40; 24.49; 24.94; 25.17; 25.28; 25.98; 30.17; 31.59; 31.77; 32.67; 33.96; 49.84; 117.01; 126.94; 127.21; 128.95; 132.14; 132.72; 134.04; 134.48; 150.47; 152.81; 158.41; 172.80; 181.79.
HRMS–C27H30N2O5 (M+Na)=485.2058 (2.3 ppm).
IR–KBr (cm-1): 3365, 2930, 2855, 1709, 1688, 990, 710
2-[(2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan3-carbonyl)amino]propanoic acid
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.46 (d, J=7.21 Hz, 3 H); 2.75 (s, 3 H); 4.46 (m, 1 H); 7.88-7.96 (m, 2 H); 8.08-8.20 (m, 2 H); 9.77 (d, J=6.85 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.73; 18.02; 48.44; 115.10; 126.33; 126.62; 127.72; 131.82; 133.21; 134.88; 135.26; 151.38; 160.71; 165.05; 173.22; 174.20; 182.77.
HRMS–C17H13NO6 (M+H)=328.0823(2.3 ppm); (M+Na)=350.0644 (2.6 ppm).
IR–KBr (cm-1): 3451, 3276, 3077, 1732, 1673, 1585, 994718.
3-methyl-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino] pentanoic acid
1HNMR (400 MHz, DMSO-d6) δ ppm: 0.95 (t, J=7.34 HZ, 3H); 1.01 (d, J=7.00Hz, 3H); 1.39 (m, 1H); 1.57 (m, 1H); 1.99 (m, 1H); 2.77 (s, 1H); 4.45 (dd, J1=8.01, J2=4.95, 1H); 7.88-7.94 (m, 2 H); 8.08-8.20 (m, 2H); 9.79 (d, J=8.07 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 12.10; 14.90; 16.30; 25.19; 37.23; 57.02; 114.98; 126.27; 126.61; 127.81; 131.77; 133.16; 134.87; 135.29; 151.43; 161.05; 165.37; 173.00; 173.22; 182.98.
HRMS–C20H19NO6 (M+H)=370.1299 (3.7 ppm); (M+Na)=392.1115 (2.8 ppm).
IR–KBr (cm-1): 3447, 3431, 3080, 1680, 1653, 993, 715.
2-[(2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan3-carbonyl)amino]-3-phenylpropanoic acid
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.70 (s, 3 H); 3.08 (dd, J1=13.94, J2=8.56 Hz, 1 H); 3.23 (dd, J1=13.88, J2=5.32 Hz, 1 H); 4.73 (liketd, J1=8.56, J2=7.98, J3=5.32 Hz, 1 H); 7.18-7.33 (m, 5 H); 7.88-7.94 (m, 2 H); 8.06-8.2 (m, 2 H); 9.83 (d, J=7.96 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.79; 37.65; 54.34; 114.86; 126.10; 126.62; 127.11; 127.67; 128.78(X2); 129.66(x2); 131.80; 133.11; 134.87; 135.27; 137.47; 151.42; 160.91; 165.24; 172.93; 173.14; 182.64.
HRMS–C23H17NO6 (M+H)=404.1140 (2.8 ppm); (M+Na)=426.0956 (1.9 ppm).
IR–KBr (cm-1): 3462, 3266, 3065, 1740, 1680, 1645, 993, 709.
3-methyl-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino]butanoic acid
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.02 (d, J=6.85 Hz, 3 H); 1.07 (d, J=6.97 Hz, 3 H); 2.27 (liketd, J1=6.85Hz, J2=6.97 Hz, J3=5.01 Hz, 1 H); 2.77 (s, 1 H); 4.44 (dd, J1=8.07, J2=4.89 Hz, 1 H); 7.87-7.95 (m, 2 H) 8.08-8.20 (m, 2 H); 9.77 (d, J=8.07 Hz, 1 H)
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.90; 18.17; 19.74; 30.46; 57.89; 114.98; 126.28; 126.62; 127.78; 131.77; 133.16; 134.87; 135.29; 151.43; 161.17; 165.37; 173.07; 173.21; 182.98; 207.50.
HRMS–C19H17NO6 (M+H)=356.1142 (3.8 ppm); (M+Na)=378.0960 (3.2 ppm).
IR–KBr (cm-1): 3447, 3265, 3080, 2967, 1740, 1679, 1592, 993, 716.
2-[(2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan3-carbonyl)amino]-4-methylsulfanyl)butanoic acid
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.99-2.09 (m, 1 H); 2.10 (s, 3 H); 2.11-2.19 (m, 1 H); 2.64 (t, J=7.52 Hz, 2 H); 2.74 (s, 3 H); 4.59 (liketd, J1=J2=7.98, J3=4.83 Hz, 1 H); 7.89-7.95 (m, 2 H); 8.09-8.20 (m, 2 H); 9.69 (d, J=7.46 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.64; 15.06; 29.97; 31.53; 51.78; 115.21; 126.54; 126.63; 127.72; 131.79; 133.22; 134.87; 135.23; 151.29; 161.06; 164.81; 173.19; 173.32; 182.66.
HRMS–C19H17NO6 S (M+H)=388.0851 (0.5 ppm); (M+Na)=410.0668 (0.2 ppm).
IR–KBr (cm-1): 3509, 3289, 3075, 2923, 1721, 1675, 1642, 1577, 1548, 995, 718
5-amino-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino]-5- oxopentanoic acid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.89-2.03 (m, 1 H); 2.08-2.17 (m, 1 H); 2.20-2.33 (m, 2 H); 2.76 (s, 1H); 4.47 (liketd, J1=J2=7.73 Hz, J3=5.44 Hz, 1 H) 6.81 (br s, 1 H) 7.33 (br s, 1 H) 7.87-7.96 (m, 2 H); 8.08-8.15 (m, 1 H); 8.16-8.22 (m, 1 H); 9.75 (d, J=7.46 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.75; 27.74; 31.54; 52.32; 115.11; 126.38; 126.63; 127.80; 131.79; 133.18; 134.87; 135.28; 151.35; 160.95; 165.08; 173.19; 173.43; 173.59; 182.75.
HRMS–C19H16N2O7 (M+H)=385.1034 (1.1 ppm); (M+Na)=407.0851 (0.3 ppm).
IR–KBr (cm-1): 3428, 3230, 3095, 1717, 1674, 1634, 1559, 990, 717.
6-amino-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino]hexanoic acid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.46-1.54 (m, 2 H); 1.59-1.68 (m, 2 H); 1.75-1.85 (m, 1 H) 1.86-1.96 (m, 1 H); 2.75 (s, 3 H); 2.82 (br t, J=7.46 Hz, 2 H); 4.46 (liketd, J1=J2=7.70 Hz, J3=5.26 Hz, 1 H); 7.90-7.94 (m, 2 H) 8.07-8.11 (m, 1 H) 8.13- 8.17 (m, 1 H); 9.74 (d, J=7.46 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.73; 22.69; 27.11; 31.38; 39.11; 52.44; 115.08; 126.33; 126.64; 127.72; 131.74; 133.12; 134.86; 135.30; 151.33; 160.93; 165.07; 173.13; 173.51; 182.73.
HRMS–C20H20N2O6 (M+H)=385.1406 (3.0 ppm).
IR–KBr (cm-1): 3440, 3273, 3071, 2945, 1691, 1578, 1553, 992, 715.
3-(4-hydroxyphenyl)-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino]propanoic acid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.72 (s, 3 H); 2.96 (dd, J1=14.00Hz, J2=8.50 Hz, 1 H); 3.11 (dd, J1=14.00 Hz, J2=5.20 Hz, 1 H); 4.63 (liketd, J1=J2=7.92 Hz, J3=5.32 Hz, 1 H); 6.60-6.69 (m, 2 H); 7.04-7.12 (m, 2 H); 7.86-7.97 (m, 2 H); 8.06-8.13 (m, 1 H) 8.13-8.25 (m, 1 H); 9.22 (br s, 1 H); 9.80 (d, J=7.46 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.24; 36.33; 54.10; 54.80; 114.29; 114.97; 125.51; 126.03; 126.85; 127.10; 130.01; 131.22; 132.56; 134.28; 134.69; 150.84; 155.92; 160.29; 164.71; 172.48; 172.57; 182.08.
HRMS–C23H17NO7 (M+H)=420.1073 (1.2 ppm); (M+Na)=442.0893 (0.8 ppm).
IR–KBr (cm-1): 3420, 3250, 3067, 1735, 1666, 1585, 993, 715.
2-[(2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan3-carbonyl)amino]pentanedioic acid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.91-2.04 (m, 1 H); 2.10-2.21 (m, 1 H); 2.37-2.46 (m, 2 H); 2.74 (s, 3 H); 4.52 (liketd, J1=J2=7.95, J3=5.38 Hz, 1 H); 7.87-7.95 (m, 2 H); 8.09-8.13 (m, 1 H); 8.15-8.20 (m, 1 H); 9.69 (d, J1=7.58 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.09; 26.64; 29.82; 51.40; 114.56; 125.92; 126.05; 127.18; 131.20; 132.62; 134.29; 134.66; 150.71; 160.45; 164.36; 172.61; 172.72; 173.49; 182.09.
HRMS–C19H15NO8 (M+H)=386.0873 (0.7 ppm); (M+Na)=408.0689 (0.2).
IR–KBr (cm-1): 3465, 3420, 3281, 3070, 2945, 1730, 1670, 1650, 994, 717.
3-h ydroxy-2-[(2-methyl-4,9-dioxo-4,9- dihydronaphtho[2,3-b]furan-3-carbonyl)amino]propanoic acid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.78 (s, 3 H); 3.77 (dd, J1=10.82, J2=3.97 Hz, 1 H); 3.90 (dd, J1=10.82, J2=4.46 Hz, 1 H); 4.53-4.57 (m, 1 H); 7.89-7.95 (m, 2 H); 8.08-8.19 (m, 2 H); 9.98 (d, J1=7.58 Hz, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.51; 54.82; 61.39; 114.55; 125.75; 126.14; 127.23; 131.34; 132.70; 134.43; 134.76; 150.92; 160.49; 165.05; 171.69; 172.77; 182.13.
HRMS–C17H13NO7 (M+H)=344.0772 (2.0 ppm); (M+Na)=366.0589 (1.3 ppm).
IR–KBr (cm-1): 3425, 3220, 3073, 3026, 1768, 1674, 1641, 1587, 1563, 990, 715.
N-{3-[4-(2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b] furan-3-carbonyl)aminobutyl]aminopropyl}-2-methyl-4,9-dioxo-4,9-dihydronaphtho[2,3-b]furan-3-carboxamide
1H NMR (400 MHz, DMSO-d6) δ ppm: 1.65-1.73 (m, 2 H); 1.74-1.83 (m, 2 H); 1.90-2.01 (m, 2 H); 2.68 (s, 3 H); 2.69 (s, 3H); 3.02-3.15 (m, 4 H); 3.38-3.48 (m, 4 H); 7.84-7.93 (m, 4 H); 8.02-8.10 (m, 4 H); 8.48 (br s, 1 H); 8.46-8.51 (m, 1 H); 9.18- 9.30 (m, 2 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 13.80; 13.93; 22.88; 25.73; 25.90; 35.93; 38.06; 44.36; 46.33; 115.05; 115.07; 125.86; 126.05; 126.09; 126.84; 126.92; 131.07; 131.10; 132.45; 134.27; 134.71; 150.41; 150.48; 160.41; 160.82; 163.44; 163.73; 172.34; 172.36; 181.68; 181.91.
HRMS–C35H31N3O8 (M+H)=622.2187 (0.5 ppm).
IR–KBr (cm-1): 3385, 3308, 3101, 1677, 1653, 849, 719.
We would like to thank the financial support of the agencies CNPq, FAPEMIG and the Oswaldo Cruz Foundation.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Siqueira EP, Ceravolo IP, Kohlhoff M, Krettli AU, Zani CL (2018) Synthesis and Antiplasmodial Activity of 2-Methyl-3- Carboxyl-Naphtho [2, 3-B] Furan Quinone Derivatives. J Med Chem Drug Des 1(2): dx.doi.org/10.16966/2578-9589.108
Copyright: © 2018 Siqueira EP, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access