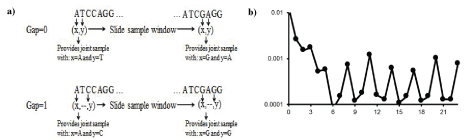
Figure 1: Codon structure is revealed in the V. cholera genome by mutual information [13] between nucleotides in the genomic sequence when evaluated for different gap sizes.

Stephen Winters-Hilt1,2* Johnathan Evanilla1,2
1Connecticut College, Department of Computer Science, New London, CT 06320, USA*Corresponding author: Stephen Winters-Hilt, Departments of Biology and Computer Science, 270 Mohegan Ave, New London, CT 06320, USA, E-mail: swinters@conncoll.edu
Many current fishery stock assessment methods strongly rely on the amount of fish harvest reported at the dock by fishermen. We seek a method for fish stock assessment that is based on transcriptome measures. In this study we were interested in the correlation between transcriptome level diversity and changes in the phenotype expression ability of commercially targeted fish. By analyzing the complexity of miRNA/RNAi 7mer binding sites in the 3’UTR regions, inferences are made as to the accessible repertoire of phenotypes for the organism. If fewer phenotypes are available, for use in response to environmental change, or for use in extending habitable niche, such as by ‘schooling’, then significant loss of fishery stock may result. Preliminary results indicate Gadus Morhua (Atlantic Cod) has undergone such a loss in transcript regulatory complexity, which appears to be associated with the collapse of the Cod fishery in the Gulf of Maine.
Fish stock diversity; Transcriptome measures; miRNA Trans-Regulation Profiling
Studies of individual regulatory elements in a variety of species [1- 3] have demonstrated the prevalence of functional motif conservation without sequence conservation. This would indicate that the sequence meta statistics, such as on distributions of anomalous regulatory motif counts, might remain the same, while the individual sub-sequences with anomalously high counts, for example, might be significantly changed from one species to the next. Where strong sequence conservation does hold, there is often associated some constraint on the encoding that prevents neutral drift to another motif sequence (such as with the overlap encoding regions described in [4]).
Cleavage stimulatory factor (CstF), is a 200 kDA heterotrimeric protein which assembles onto the 3’ end of a pre-mRNA (probably as a dimer). CstF binding promotes the polyadenylation process. Once polyadenylated, the mature mRNA is ready for export outside the nucleus. Not surprisingly, the amount of CstF depends on cell cycle [5]. CstF is also known to play an active role in response to DNA damage [6], where it has been found that cells with lower levels of CstF have less viability for survival following UV exposure. CstF is seen to play a critical role in tumor cells as well. Many tumors have been found to have a mutated p53 gene (the most commonly mutated gene in human tumors). Recent studies of p53 show that it inhibits mRNA 3’ processing via interaction with CstF [7]. P53 is also known to transactivate miRNAs, allowing large changes in expression for miRNA targeted genes in later post-translational processing [7]. P53 and CstF together are at the nexus of a critical regulatory control via 3’ processing. Not surprisingly, as we will show, the motif ‘footprints’ of the CstF binding site are one of the most statistically strong motifs (high count anomalous) in the 3’ region of mRNAs. The prevalence of the CstF motif seen in the ‘healthy’ species is found to be reduced and less varied in damaged fish stocks (as will be shown), and is associated with reduced, less targeted, CstF binding.
Transcriptome-wide comparisons have been done via SNP profiling, where identification and use of SNP markers permit a fine-scale stock identification and tracking, and could eventually allow a deeper understanding of ecotype divergence [8]. In a study of pacific herring [9] almost 11,000 potential SNPs were identified, of which 96 were directly tested. Of those 96, six were found to provide excellent sub-population biomarkers. SNP discovery is more scalable than SNP validation. SNP validation is inherently more difficult than motif validation in that the single nucleotide has no additional implicit information than the ‘one bit’ of information typically encoded in a two-state SNP. A motif that is 10 bases long, on the other hand, has 4 ^ 10=2 ^ 20~=10 ^ 6 possibilities, of which some can occur with anomalously high counts, allowing for six orders of magnitude greater internal or ‘implicit’ information content. This allows a preliminary validation process to be done much more in the computational (scalable) realm, if not entirely computational if referring to a meta-level statistical analysis as we will be being done here.
We describe an investigation into transcriptome diversity, and associated phenotype expression ability, of commercially targeted fish. This is done by analyzing the complexity of miRNA/RNAi 7mer-based regulatory motif footprints in the 3’ untranslated region (3’UTR) of protein coding transcripts. There appears to be a ‘normal’ 7mer count distribution profile. The hypothesis is that a reduction (or significant deviation from normal) in these motif footprints correlates with loss of transcriptome diversity and a less abundant stock.
The transcriptome/EST data analysis is done using on ORF-finder program written in Perl [4]. EST 3’UTRs are identified, wherein anomalously recurring 7-base sequences, known as “7mers,” are sought. By analyzing the distribution on 7mers, a crude assessment of transcriptome regulatory diversity is inferred, with possible implications for fish stock assessments.
In this paper we perform transcriptome-wide studies, where we do transcript fingerprinting not via a SNP profile on each transcript [10], but via a miRNA binding site (7mer) profile on each transcript’s 3’UTR region. By doing a meta statistics analysis on the anomalous motif occurrences we will show evidence of significant trans-regulatory damage in Atlantic Cod (G. morhua) which is known to be in an overfished status where overfishing is still occurring [11].
In Current fish stock assessment methods Section, a description is given of the current fish stock assessment methods. In Entropy measures, statistical linkage, and mutual information: Codon & ORF discovery Section, bioinformatics methods are used to quickly rediscover ORF structure from raw genomic data (to prove their utility) then the methods are used to identify the anomalously high-count sub-sequences found before and after the protein coding regions (the cis-regulatory and transregulatory motifs (where cis motifs are ‘upstream’ of coding, i.e., they are the sequence region to the left of the coding sequence region, while trans is to the right of the coding region).
Fisheries stock assessment refers to the analysis of the past and current status of a group of fish that live in the same geographic area, in order to learn more about the effects of fishing and other factors. The information obtained from stock assessments helps fisheries managers make sustainable decisions.
Stock assessments are done using models which rely on three different types of data: catch, abundance and biology. Catch data is simply the amount of fish taken from a stock of fish by fishing. There are many ways fisheries managers can obtain this data, including dockside monitoring, logbooks from commercial fishermen, observers that go to sea with commercial fishermen, and sampling the catch of recreational anglers. Abundance data is a measure or representation of the amount of fish that are actually in the stock. This type of information usually is generated by a statistical model which analyzes sampling data from fishery-independent surveys. These surveys take place on research vessels or contracted fishing vessels and use standardized sampling methods. Biology data adds the aspect of individual fish growth and mortality into the model. Some aspects of biological data that are incorporated can include growth rates, reproductive rates and movement.
The models which are used to conduct stock assessment differ among different commercial fisheries, and are limited by the amount and type of data available to use. Many other factors are also often incorporated into these models. A species’ position in its larger food web, competition between other species, habitat and physical environmental conditions are all other aspects that can be taken into account. While some fisheries are very well maintained, others may need some work to better the way in which they are maintained.
The degree of randomness in a discrete probability distribution P is measured in terms of Shannon entropy [12]:
S(P) = Σk pk log(pk )
where P has outcome probabilities {pk }.
When comparing discrete probability distributions P and Q, both referring to the same N outcomes, the proper measure of their difference is measured in terms of their (possibly symmetrized) relative entropy [12] (a.k.a. Kullback-Leibler Divergence):
D(P||Q) = Σk pk log(pk /qk )
where P and Q have outcome probabilities {pk } and {qk }.
In evaluating if there is a statistical linkage between two events X and Y we are essentially asking if the probability of those events are independent, e.g., does P(X,Y)=P(X)P(Y)? Since this reduces to measuring the difference between two probability distributions: P(X,Y) and Q(X,Y)=P(X)P(Y), the relative entropy between P and Q is sought, where D(P(X,Y) || P(X)P(Y) )is the definition of ‘mutual information between {X,Y}: MI(X,Y)=D(P(X,Y) || P(X)P(Y)).
Mutual information allows statistical linkages to be discovered that are not otherwise apparent. Consider the mutual information between nucleotides in genomic data when different gap sizes are considered between the nucleotides as shown in figure 1a. When the MI for different gap sizes is evaluated (Figure 1b), a highly anomalous long-range statistical linkage is seen, consistent with a three-element encoding scheme (the codon structure is thereby revealed)
Figure 1: Codon structure is revealed in the V. cholera genome by mutual information [13] between nucleotides in the genomic sequence when evaluated for different gap sizes.
Once codon groupings are revealed, a frequency analysis on codons can be done, and the ‘stop’ codons are found to be rare. Focusing on the stop codons it is easily found that the gaps between stop codons can be quite anomalous compared to the gaps between other codons. Open reading frames (ORFs) are regions that have no stop codon {(uaa),(uag),(uga)} when traversing with a particular codon framing. The restriction to larger ORFs is due to their highly anomalous occurrences and likely biological encoding origin, e.g., the long ORFs give a strong indication of containing the coding region of a gene. By restricting to transcripts with ORFs ≥ 300 in length, we have a resulting pool of transcripts that are mostly true coding transcripts.
Once the anomalous ORF structure is identified, nearby associated encoding anomalies are discovered (which in turn serve as validators), such as transcription start site recognition, in case of genomic sequence, or start/end of coding region recognition, in case of genomic or transcriptomic sequence information. The cis- and trans-regulatory regions are shown in figure 2, with cis-regulation via protein transcription factors dominating for DNA mRNA regulation, and miRNA template strand recognition (via RNAi) regulation dominating mRNA protein processing.
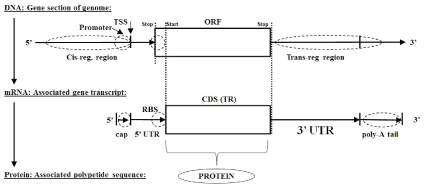
Figure 2: Classic (monocistronic) central dogma: DNA mRNA Protein
A transcriptome-wide study is done on numerous species of fish (details to follow in Methods and Results). For a given species, the length distribution on their 3’UTR regions is examined, with specific plots shown for three species of fish in the Methods, where the selection of >300 ORF and >200 3’UTR is made in the initial data handling (as summarized in table 1 in the Methods to follow).
| Species | GenbankESTs | uniq_ORF ≥ 300 | 3UTR ≥ 200 |
| Tuna | 10,163 | 5,366 |
1,739 |
| Salmon | 498,523 | 232,014 | 96,084 |
| Cod | 257,255 | 117,443 | 41,673 |
| Catfish | 139,475 | 60,094 | 24,558 |
| Pufferfish | 26,069 | 11,274 |
2,599 |
| Cyprinus | 47,738 | 26,579 | 10,166 |
| Dicentrarchus | 55,837 | 25,929 | 9,904 |
| Disso | 37,104 | 17,371 | 4,803 |
| Hippoglossus | 20,836 | 15,066 | 5,659 |
| Osmerus | 36,788 | 28,693 | 16,040 |
| Sparus | 29,216 | 38,034 | 8,710 |
| Zebrafish | 1,488,339* | 121,554 | 44,253 |
| Astyanax | 189,864 | 118,036 | 43,094 |
Table 1: Preprocessing of mRNA/ESTs unique strands with ORFs ≥ 300 also with 3UTRs ≥ 200.
*first 20% of genbank sequences for zebrafish.
The analysis in this paper focuses on data presented at the transcriptome level, particularly that from EST processing. This allows analysis to be done at the earliest opportunity since EST generation is an essential first step in genome construction, SNP discovery, and microarray design. Assuming the collection of transcripts has already been filtered such that each transcript has at least one ORF length greater than or equal to 300 nucleotides, we now filter further according to retaining those transcripts with 3’UTR regions 200 nucleotides in length or greater (Figure 3), with results as shown in table 1.
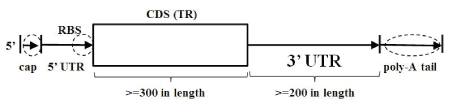
Figure 3: Transcript selection: ≥ 300 length ORF region and ≥ 200length 3’UTR region.
Referring to Salmon from table 1 as an example: there are 498,523 EST transcripts from Genbank that are validated via a high-confidence BLAST score alignment to a Genbank-annotated protein coding mRNA. These EST transcripts are scanned with six ORF-finder passes: three ORF passes in the forward direction, for the three positive strand ORF framepasses, and three ORF frame-passes on the reverse-compliment strand for the negative DNA strand genes. (There are three frame passes because the codon encoding element is three bases long, such that a tiling over the sequence with codons is possible with three different codon ‘frame’ conventions). We restrict to transcripts for which at least one ORF ≥ 300 bases in length is found according to any of the six aforementioned framepasses. Of the ORF ≥ 300 sequence, we restrict further to those having 3’UTR regions greater than 200 bases.
The cutoff of ≥ 200 3’UTR length is justified on a similar basis to the ORF cut-off that is typically used (mentioned earlier). As with the ORF length distribution, the 3’UTR distributions reveal a clear deviation from geometric fall-off on length (as might be expected from a random process), and if sufficiently far into the heavy tail region (with non-zero counts), where the geometric distribution fit would indicate a zero count, then all such instances have a high likelihood of pertaining to a biological encoding. The 3’UTR length histograms for three species of fish are shown in figure 4.
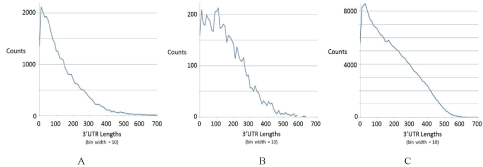
Figure 4: 3’UTR Length Distribution Profiling/Validation. Length distribution on 3’ UTR regions for tuna, salmon, cod (from A-C).
In each instance in figure 4, a fit to a geometric distribution can be based on the short 3’UTR lengths (just as with short ORF lengths) to estimate the random approximately geometric distribution, from which the deviation of the actual length distribution is can be estimated. For the species shown in figure 4 and also listed in table 1, the deviation is notable for lengths ≥ 200, thus the choice of cut-off. What is perhaps even more notable is that species-wide uniformity in the maximal 3’UTR lengths. Notice in figure 4 that there are no 3’UTR regions greater than 600 bases, with very few greater than 400 bases. The same is also found to hold for the other fish in table 1, and for human, moue and a number of other organisms (not shown). A heavy tail 3’UTR distribution with strict fall-off to zero at 600 length or longer serves as a further validation on acquisition as well, since it appears to be a universal.
In the Methods we describe how Genbank mRNA/EST data is downloaded, filtered, and rudimentary validation is done. In this process all of the fish 3’UTR regions shared similar meta-statistical features as already mentioned. In what table 2 is shown further transcriptomewide processing for the fish species described in table 1. The first column describes the transcripts obtained after the aforementioned ORF ≥ 300 and 3’UTR ≥ 200 filters, plus the added filter of requiring that the first 35 bases in a 3’UTR region be unique (otherwise take the longer transcript and discard the other). The transcripts meeting the various filters indicated are then passed through a prokaryotic gene-finding program that does three ORF passes in the forward direction then three ORF passes on the reverse complement read of the sequence. The six ORF passes filter according to the ORF ≥ 300, 3’UTR ≥ 200 and ‘35uniq’, and their overlap topology is noted as done in previous work [13]. If a transcript has both forward and reverse encoding, each of which meets the strict filtering criteria (ORF ≥ 300, etc.) then the transcript is referred to as ‘dual;’ in table 2. The extent of dual encoding revealed at this stage of the transcriptome-wide validation process was a surprising result and is described further in the study by Winters-Hilt S (2017) [4], so won’t be discussed further here, other than to point out a universal amount of ‘duality’ appears to occur in the 7%-15% range (and this is seen to hold for human and mouse and other transcriptomes as well). The amount of same read direction overlap encoding is also significant, and also typically falls in a range (between 11% and 18%) that can serve to validate acquisition.
| Species | # of Genbank mRNA/EST sequences with ORF ≥ 300, 3UTR ≥ 00, & uniq35start | % column 1 mRNA/ EST sequences that are dual | % ORFs from column 1 sequences that are in (loosely filtered) operons | % ORFs from column 1 overlapping with same read direction: |
| Bluefin Tuna Thunnus thynnus |
1541 | 9.5 | 0.63 | 11.8 |
| Atlantic Salmon Salmo Salar | 82007 | 8.0 | 0.86 | 13.5 |
| Atlantic Cod Gadus Morhua | 34069 | 10.1 | 1.17 | 17.0 |
| Blue Catfish Ictalurus Furcatus |
20727 | 8.7 | 2.06 | 13.7 |
| Japanese Pufferfish Takifugu Rubripes |
2313 | 6.5 | 0.19 | 12.2 |
| Carp Cyprinus Carpio | 8275 | 12.4 | 1.50 | 14.6 |
| European Bass Dicentrarchus Labrax | 8372 | 9.8 | 0.97 | 13.1 |
| Antarctic Toothfish Dissostichus mawsoni |
4151 | 7.1 | 0.40 | 14.2 |
| Atlantic Halibut H. Hippoglossus |
4579 | 10.9 | 0.51 | 14.7 |
| Rainbow Smelt Osmerus Mordax | 12409 | 14.3 | 2.03 | 17.9 |
| Gilt-head Bream Sparus Aurata | 13830 | 9.8 | 1.15 | 12.7 |
| Zebrafish Danio Rerio |
37844 | 7.4 | 0.62 | 13.9 |
| Blind Cave Fish Astyanax Mexicanus | 37,695 | 7.2 | 0.23 | 12.8 |
Table 2: 3’UTR Sample Selection and associated ORF topology. The number of ESTs used in the transcriptome analysis and their ORF topology
Perhaps the most concerning 3’UTR acquisition validation statistic in table 2 is the percentage of ORFs recognized as being part of an operon. As mentioned in the methods, there is no direct handling on operon structure (if present) with the simple algorithm used. Rather, operon handling is done via the iterative bootstrap process mentioned earlier. In the fish analysis a crude operon recognition was done for any transcript that had multiple ORFs non-overlapping, where those ORFs would all be considered part of a single operon, for which a single 3’UTR region is indicated (to the right of the rightmost ORF in the operon). An operon is a cluster of coding regions under common cis-regulation, where the ORFs enclosing those coding regions may overlap to a small extent, such that the operon construction algorithm based on sets of disjoint ORFs (with results shown in table 3) only captures part of the operon structure (providing an estimate). In practice, tuning on allowed overlap amounts reveals an upper bound on percentage of operon structure that is roughly twice that shown in table 2, for most species, but less than 3% for all. Since the upper bound on operon structure is 3% of the filtered data obtained thus far, this means that we have at most a 3% source of count errors in the 3’UTR 7mer motif analysis. This level of error can be tolerated with the motif-type signal analysis that follows, given the cutoffs that are employed, so further efforts to deal with the operons will be left to when it is necessary.
| Species /7mer_counts | µ(mean) | o (std.dev) | σ/µ | #> µ + 3σ | #> µ +1σ | % ‘> µ + 1σ’ with no4 |
7A-mer counts | #7A/µ |
| Bluefin Tuna Thunnusthynnus |
30.6 | 22.87 | 0.745 | 177 | 2005 | 42.3 | 920 | 30 |
| Atlantic Salmon SalmoSalar | 1820 | 1280 | 0.703 | 172 | 2211 | 45.7 | 28940 | 16 |
| Atlantic Cod GadusMorhua | 794 | 919 | 1.157 | 70 | 767 | 15.0 | 88430 | 111 |
| Blue Catfish IctalurusFurcatus |
478 | 442 | 0.925 | 107 | 1348 | 32.3 | 30647 | 64 |
| Japanese Pufferfish TakifuguRubripes |
43 | 38 | 0.883 | 247 | 1673 | 55.9 | 1796 | 42 |
| Carp CyprinusCarpio | 202 | 170 | 0.842 | 114 | 1684 | 40.6 | 12078 | 60 |
| European Bass DicentrarchusLabrax | 190 | 152 | 0.800 | 143 | 2047 | 44.9 | 6152 | 32 |
| Antarctic Toothfish Dissostichusmawsoni |
86.5 | 87.7 | 1.014 | 118 | 1497 | 38.3 | 6830 | 79 |
| Atlantic Halibut H. Hippoglossus |
104.1 | 72.8 | 0.699 | 233 | 2320 | 58.6 | 913 | 9 |
| Rainbow Smelt OsmerusMordax | 348.6 | 234.0 | 0.671 | 191 | 2238 | 47.1 | 3554 | 10 |
| Gilt-head Bream SparusAurata | 329.6 | 308.3 | 0.935 | 107 | 1628 | 42.1 | 22283 | 68 |
| Zebrafish Danio Rerio |
816 | 1249 | 1.531 | 61 | 652 | 28.4 | 133791 | 164 |
| Blind Cave Fish AstyanaxMexicanus | 753.0 | 680.5 | 0.904 | 185 | 1778 | 44.3 | 26716 | 35 |
Table 3: 7mer count statistics. Noisy ESTs show as significant over-counting in ‘aaaaaaa’ 7mers, which, via #polyA/mu, is used as a gauge of the noise in the dataset in the table.
At this point we have a set of transcriptome-wide 3’UTR extracts for several species of fish that is highly vetted. Let’s now examine these sets of 3’UTR regions for their 7mer count statistics at a meta-statistical level (Table 3), without reference to specific sequence information, and then at a direct statistical level as relates to particular signaling motifs that have been identified. In table 3 it is shown that the transcriptome-wide 3’UTR 7mer count statistics, including the mean count and standard deviation on counts, etc., for each species.
If σ/µ<1.0 we have more of a Gaussian structure emerging for k-mer count distributions, with easily identifiable “heavy-tail” statistical anomalies, while σ/µ>1.0 indicates a more uniform distribution. The σ/µ>1.0 of the Cod 7mer distribution is partly an artifact of the high poly-A 7mer counts distorting the count statistics, however, as other species transcriptome data with σ/µ>1.0 also had high #7A/u. So σ/µ>1.0 is not a distinguishing characteristic. If we look further at the types of motifs, however, we find that the high-count 7mers typically fall into two categories: 4 or more bases the same, or no more than 3 bases the same (“no4”). If we consider the percentage of high-count anomalous 7mer with no more than 3 bases of the same type we see that Atlantic Cod is singled out. If we look further, into the list of high-count sequences we see that there is a group of 4-or-mor-bases-the-same motifs missing as well, many of them variants of the CstF motif. Thus Atlantic Cod has a notably reduced TF binding site strength for CstF and is lacking a large number of “no4” 7mer miRNA targets. This is discussed further in the Discussion, but the main result is seen here in the statistics. In these results we are seeking a trans-regulation diversity biomarker (that is meta-statistics based) and the no4 statistic appears to suffice in this role by singling out atlantic cod where fishery collapse has occurred from numerous other species not suffering from such as drastic niche failure.
To recap, first recall the typical eukaryotic 3’UTR signaling (starting with the stop codon at the left):
---|TAA-------(T-rich)-----(*)-----AATAAA-----(poly-A site)----(T/ GT rich)----
So, we expect to see in the list of most frequent 7mers in the 3’UTR:
(1) 7mers that are T-rich: ttttttt, ttatttt, tttattt, etc.
(2) 7mers that are A-rich and poly-A with very high counts,
(3) 7mers that have ‘AATAAA’
(4) 7mers that are GT-rich for alt-polyA via (*)=(GT rich) signal
All of which is seen. (Note how all of the 3’UTR signaling related to mRNA production processing have multi-target repeat type signals.)
Atlantic Cod, however, is found to have significantly less ‘diffuse GT’ motif than other species of fish (not shown), the motif involved in CstF recruitment and related poly-A cleavage site selection: e.g., g(tg)(tg)(tg) motifs are seen in cod, but not c(tg)c(tg) or c(tg)tc(tg). Damaged CstF activity is associated with disease and enhanced (detrimental) sensitivity to environmental stimulus – yeast cells with reduced levels of CstF display an enhanced sensitivity to UV treatment, for example.
We expect to see 7mers with high frequencies when they associate with miRNA binding sites. It is known that many miRNA 7mer binding sites are controlled with high-specificity (i.e., the 7mer-target has no repeating elements that would allow multiple targeting miRNAs), while other miRNA targeting is meant for multiple binding sites (with 7mer binding sites with repeats). We can ‘lock’ onto the high-specificity miRNA signaling by focusing on 7mers with low motif-pattern repetition – this is accomplished by focusing on 7mers that have no more than three bases of the same type (the ‘no4’ 7mers).The notably less informed (Shannon entropy greater) 7mer count distribution for Cod is hypothesized to relate to a reduced complexity in 7mer-based miRNA/RNAi regulatory capabilities.
If Cod has less trans-regulatory capabilities, resulting in a less diverse selection of phenotypes needed in order to robustly respond to environmental change, then it will become endangered as a species from much more minor environmental changes, as appears to be the case since the collapse of the Cod fisheries in the Northeast. The loss of trans-regulatory diversity may provide a new indicator of overfishing and environmental strain (due to shift in feeding areas further from spawning areas for example), and may provide an early transcriptome-based indicator of fishing stock damage for commercial fisheries.
Atlantic Cod appears to have significantly less ‘diffuse GT’ motif in its 3’UTR transcripts, indicative of compromised CstF recruitment. Damaged CstF activity is associated with disease and enhanced (detrimental) sensitivity to environmental stimulus – enhanced sensitivity to UV for example. Atlantic Cod also appears to have significantly less trans-regulatory high-specificity (‘no4’) miRNA complexity than other fish. Less trans-regulatory complexity will lead to less diverse mRNA trans-regulation control of phenotypes, leading to less robust response to environmental change. These results identify a meta-statistical transcriptome-based stock assessment biomarker for potential or occurring ecotype collapse. The biomarker correctly identifies Atlantic Cod as a species at risk from a set including twelve other fish species not thought to be at risk.
The authors would like to thank Connecticut College and QLS for research support.
Download Provisional PDF Here
Article Type: Research Article
Citation: Winters-Hilt S, Evanilla J (2017) Characterization of Fish Stock Diversity via ESTBased miRNA Trans-Regulation Profiling. Int J Mol Genet Gene Ther 3(1): doi http://dx.doi. org/10.16966/2471-4968.110
Copyright: © 2017 Winters-Hilt S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access