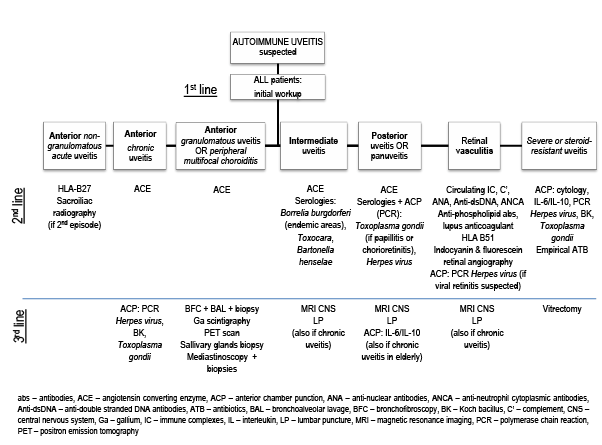
Figure 1. Algorithm for the diagnosis of a suspected autoimmune uveitis [3,7,20].
Inês Chora*# Tiago Borges# Carla Dias Carolina Ourique João Vilaça Sara Ferreira Paula Vaz-Marques Sérgio Silva Carlos Dias
Department of Internal Medicine, São João Hospital Center, Al. Prof. Hernâni Monteiro, 4200-319 Porto, Portugal*Corresponding author: Inês Chora, Department of Internal Medicine, São João Hospital Center, Al. Prof. Hernâni Monteiro, 4200-319 Porto, Portugal, E-mail: i_chora@yahoo.com
Uveitis is an intraocular inflammation of multiple possible etiologies, making its clinical management particularly challenging. Autoimmune uveitis is a leading and under-recognized cause of preventable blindness. Systemic sarcoidosis, spondyloarthritis, Behçet’s syndrome and VogtKoyanagi-Harada’s syndrome are systemic autoimmune diseases frequently associated with uveitis.
A stepwise approach of autoimmune uveitis is essential to appropriate diagnosis and early treatment. When autoimmune uveitis is suspected, patients should be promptly referred to an ophthalmologist for the classification of ocular lesions; thereafter, a multidisciplinary investigation of a subjacent systemic disease must follow. Currently, no consensus diagnostic algorithm is accepted for autoimmune uveitis and an individualized approach is generally used. The treatment of autoimmune uveitis depends on ocular semiology, being further refined if an underlying systemic disorder is identified.
This review focuses on relevant clinical, diagnostic and therapeutic issues related to autoimmune uveitis in adults, with emphasis on the main associated systemic autoimmune diseases. A diagnostic algorithm for autoimmune uveitis is proposed.
Autoimmune uveitis; Behçet’s syndrome; Sarcoidosis; Spondyloarthritis; Vogt-Koyanagi-Harada syndrome
The eye has a particular relationship with the immune system which is known as “immune privilege” and involves physical barriers, immunosuppressive factors and protein antigens [1]. The term “uveitis” is often used to describe the inflammation of the middle layer of the eye, known as uveal tract, but in general it can be used to describe any intraocular inflammatory process [2,3]. The International Uveitis Study Group classification system considers location, clinical course (acute if lasting less than three months, chronic if otherwise, and recurrent when acute flares appear after a complete resolution of the previous event) and laterality (unilateral or bilateral in relation to both eyes) to define its pattern [3]. According to location, uveitis is considered to be anterior if it affects the iris or the ciliary body (iritis or iridocyclitis), intermediate when limited to the vitreous (vitritis), peripheral retina, pars plana or the ciliary body, posterior if it involves the choroid and possibly the retina (choroiditis, retinochoroiditis and chorioretinitis), and finally panuveitis when at least two of these segments are involved [3,4]. Uveitis can be a co-manifestation of systemic autoimmune rheumatic diseases (SARD), a side effect of medications, a consequence of exposure to toxins or just an idiopathic disorder [5]. It has been traditionally categorized as infectious or non-infectious [6]. Infectious uveitis can be secondary to viral, bacterial, parasitic or fungal infections [5,7]. Non-infectious uveitis is believed to be either autoimmune, when it is mediated by aberrant immune recognition of self, or immune-mediated, if an innate inflammatory reaction is triggered by environmental or autologous signals [8,9]. Some authors consider autoimmune uveitis (AIU) to be an organ-specific disorder characterized by irreversible ocular lesions, even though some SARD can present uveitis and overlapping AIU [10]. In this article, AIU is considered to include both organ-specific and SARD-associated uveitis, regardless of which immune response (adaptive or innate) predominates.
SARD like spondyloarthritis (SpA), systemic sarcoidosis (SS), Behçet’s syndrome (BS) and Vogt-Koyanagi-Harada syndrome (VKH) are common causes of uveitis in adults [8] (Table 1).
Acute recurrent unilateral anterior uveitis has been estimated to represent about one third of uveitis cases and, in about half of the cases, it is caused by SpA [3,5]. Acute non-recurrent unilateral uveitis corresponds only to a small minority of anterior uveitis cases, being idiopathic in 50% and associated with SpA in about one fifth. Acute bilateral anterior uveitis is idiopathic in almost half of the patients but can be associated with psoriasis, tubulointerstitial nephritis and uveitis syndrome (TINU) or SpA [3]. Similarly, chronic anterior uveitis (CAU) is often idiopathic but can be associated with juvenile idiopathic arthritis (JIA) in children, Sjögren syndrome, sarcoidosis or SpA [3]. Posterior uveitis and panuveitis are usually due to toxoplasmosis if associated with chorioretinitis, while posterior uveitis with retinal vasculitis (RV) and no systemic involvement is idiopathic in the majority of the patients but can associated with BS, sarcoidosis or VKH [3,5]. If panuveitis occurs along with RV, BS is likely in more than one third of cases, but idiopathic cases, inflammatory bowel disease (IBD) and sarcoidosis are also possible diagnosis, whilst panuveitis with vitritis is either idiopathic (in about two fifths of the affected patients) or secondary to SpA in a minority [3]. In contrast to other locations, intermediate uveitis is idiopathic in about three quarters of the cases, even though multiple sclerosis (MS), SpA and sarcoidosis are possible causes [3,5]. In the rare cases where exudative retinal detachment is present, VKH is the diagnosis in the great majority of the cases [3].
An epidemiological study has estimated the prevalence of uveitis in the general population to be 0.1% and the incidence 52 cases per 100 000 persons-year, affecting mainly patients between their third and fifth decades of life, with both sexes equally represented [3,15]. Anterior uveitis represents up to 90% of the cases [3]. After diabetes and glaucoma, uveitis is the most blinding entity, especially in younger patients [16]. It causes about 10% of visual losses and 5 to 20% of legal blindness cases in developed countries [3]. Globally, a specific diagnosis is possible in about 60% of cases but, in extremes of age, the diversity of specific entities as a cause of uveitis seems to be narrower and, specifically in the elderly, infections and masquerade syndromes are more common [5].
Disorder |
Prevalence of uveitis | Type of uveitis |
| Adult-onset Still’s disease | Uncommon | Usually granulomatous iridocyclitis |
| ANCA-associated vasculitis | More common in granulomatosis with polyangiitis (10-20%) | Often granulomatous anterior uveitis, but also intermediate uveitis, retinal vasculitis, chorioretinitis and florid uveitis |
| Behçet’s syndrome | At least 50% | Usually bilateral CAU, but also posterior involvement frequent |
| Cogan’s syndrome | 30% (for iridocyclitis) | Usually anterior |
| Essential mixed cryoglobulinemia | No data found | Bilateral CAU |
| HUVS | 30% | Bilateral CAU |
| IBD without arthritis | 2.5% in Crohn’s disease 5-12% in ulcerative colitis | Usually bilateral anterior (or posterior) uveitis |
| Kawasaki disease | Over 80% | Anterior uveitis |
| Juvenile idiopathic arthritis | 3 to 16% (mainly oligoarticular subtype) | CAU or AAU |
| Polyarteritis nodosa | No data found | Usually posterior |
| Relapsing polychondritis | Rare | Usually non-granulomatous, either hypopyon or non-hypopyon uveitis |
| Rheumatoid arthritis | Annedoctal reports 50% | Anterior, non-granulomatous Occurs as a complication of scleritis |
| Sarcoidosis | 10 to 20% | Usually granulomatous AAU |
| Sjögren syndrome | Rare | Bilateral, chronic, anterior and posterior uveitis |
| Spondyloarthritis Ankylosing spondylitis IBD-associated Psoriasis-associated Reactive arthritis |
25 to 40% 10 to 36% 25% 25% |
Usually AAU Usually AAU, but also chorioretinitis AAU or CAU Usually AAU |
| Systemic lupus erythematosus | 10% (symptomatic) / 29% (fluorescein angiography) |
Usually posterior |
| Systemic sclerosis | Annedoctal reports in adults | Anterior uveitis |
| TINU | Virtually all cases | Bilateral anterior non-granulomatous uveitis |
| VKH | Virtually all cases | Bilateral granulomatous panuveitis, iridocyclitis |
Table 1: Systemic autoimmune rheumatic disorders associated with uveitis* [2,4,7,8,11-14] AAU – acute anterior uveitis; ANCA – antineutrophil cytoplasmatic antibodies; CAU – chronic anterior uveitis; HUVS – hypocomplementemic urticarial vasculitis syndrome; IBD – inflammatory bowel disease; TINU – Tubulointerstitial nephritis and uveitis syndrome; VKH – Vogt-Koyanagi-Harada syndrome. * Other systemic autoimmune rheumatic disorders such as vitiligo, multiple sclerosis, autoimmune hepatitis and IgA nephropathy that are associated with uveitis are organ-specific, so they are not considered in this table, similarly to infectious, autoinflammatory and ocular syndromes.
Genetic factors play an important role in uveitis pathogenesis: major histocompatibility class I (MH1) proteins have been linked to anterior uveitis, whilst MH class II (MH2) proteins have been associated with posterior uveitis [3]. HLA-B27, HLA-DR8, HLA-B60, ‘protein tyrosine phosphatase, non-receptor type 22 (lymphoid)’ (PTNP22) and polymorphisms in ‘proteasome (prosome, macropain) subunit, beta type, 9’ (PSMB9, LMP2) gene have been linked to uveitis in certain populations [3,17]. On the other hand, environmental factors seem to be important in uveitis triggering, specifically Gram-negative infections in acute anterior uveitis (AAU), with a possible role for intestinal mucosa or its lymphoid tissue [3].
AIU is believed to be mediated by T cells and T helper 1 (Th1) and Th17 subsets have deserved special attention as pathogenic effectors, although some cytokines produced by these cells have protective effects [18]. ‘Collagen, type I, alpha 2‘ (COL1A2) has been proposed as a target in experimental AIU in rodents [4]. Interleukin 6 (IL6) is known to be in high concentrations in the vitreous fluid of chronic AIU patients, and both IL6 and ‘interleukin 23, alpha subunit p19‘ (IL23A) induce the expression of the Th17 subset [8]. Others cytokines such as ‘interferon, gamma‘ (IFNG, IFN-γ), ‘interleukin 1, beta‘ (IL1B), interleukin 17A (17A) and tumor necrosis factor (TNF, TNF-α) have proinflammatory and/or pathogenic properties in AIU models, whilst protective cytokines such as IL4, IL10 and IL22 may also be elevated [18]. IL2 is involved in conventional and regulatory T cells survival, being elevated either in aqueous humor and serum of patients with AIU [18]. Instead, innate production of IFN-γ and IL-17 may have a regulatory role in uveitis [19]. Also, regulatory mechanisms such as control of myeloid cells through receptors and soluble inhibitory factors, active immune surveillance and regulation of the blood retinal barrier appear to be relevant [6].
The clinical manifestations of AIU depend on the affected uveal region [8]. Anterior uveitis symptoms include eye redness, blurred vision, photophobia, periorbital pain, floaters and headaches, while posterior uveitis usually manifests by floaters, blurred vision, photopsia and even severe visual loss [17]. RV is a subtype of uveitic disease that commonly presents with no symptoms and may be the initial presentation of SARD [16]. Retinal vessel sheathing is the main diagnostic sign in fundoscopy, while different entities often have a tropism for different vessel types: arteries are preferentially affected in systemic lupus erythematosus (SLE), granulomatosis with polyangiitis (GPA, also known as Wegener granulomatosis) and polyarteritis nodosa (PAN), whilst veins are the main target in SS and MS [16].
The aim of this article is to review the main SARD that are associated with AIU and their clinical management in adults. Therefore, infectious uveitis, autoinflammatory disorders (such as immune recovery syndrome or familial periodic fevers), ocular syndromes (for example, birdshot choroidopathy) and organ-specific autoimmune diseases (autoimmune hepatitis, IgA nephropathy, MS) will not be approached.
General analysis |
Serology Imaging exams |
CT – computed tomography, TPHA – Treponema pallidum hemagglutination assay, VDRL – venereal disease research laboratory |
|
Table 2: Initial workup in suspected autoimmune uveitis [3,7,20].
The first step in the evaluation of uveitis consists in the prompt referral to an ophthalmologist, for the precise classification of ocular lesions and exclusion of ocular diseases. In patients presenting for the first time with isolated ocular symptoms, the role of the ophthalmologist is crucial in making a diagnosis or providing other specialists necessary information to create a focused differential diagnosis. Uveitis associated with SARD usually manifests as a single episode of acute onset affecting one eye, but in some cases may become a chronic inflammatory process. An exhaustive investigation of a subjacent systemic disease must be performed and, thereafter, treatment should be planned according to the involved organs [3,8,16,17,20,21].
A tailored approach in the hands of ophthalmologists, internists, rheumatologists or infecciologists with experience in uveitis avoids unnecessary diagnostic tests and is essential for the correct assessment and treatment of these patients. Attending to the extensive etiologic possibilities, managing uveitis is a challenge [7,20]. Even in the absence of systemic manifestations of SARD, a patient with uveitis requiring immunosuppressive therapy should be followed by a multidisciplinary team [20]. The referral of a patient with uveitis to an expert is often delayed because uveitis is commonly under-recognized, which increases the risk that uveitis will result in irreversible damage.
When approaching a patient with a suspected AIU, a detailed medical history including present illness, past medical history and a thorough review of systems, in addition to the ocular history, must be performed. The patient’s previous history of infections, tumors, drug sensitivity and rheumatologic diseases must be obtained. The ophthalmic examination must be complemented with a meticulous systemic examination [16].
The diagnosis of an AIU might be based on ophthalmological and extraophthalmological manifestations, if clinical and biological criteria of a suitable systemic disease are present. In its absence, it is necessary to review ophthalmological semiology and redirect investigation’s path. Currently, no consensus algorithm is accepted or validated for the diagnosis of AIU [7,20]. An individualized and pluridisciplinary approach, taking into account anatomical and clinical characteristics of ocular lesions, as well as extra-ophthalmological manifestations, is generally used. When this first approach raises the suspicion of an underlying SARD causing uveitis, every patient must undergo an initial diagnostic workup, as presented in Table 2. Further investigations may be planned thereafter (Figure 1).
Treponemal tests and chest radiography are recommended because syphilis and sarcoidosis do not follow any characteristic pattern [3]. An algorithm for the clinical diagnosis of a suspected AIU in the adult patient is proposed in Figure 1.
In immunocompromised patients, the above mentioned infectious causes of uveitis must be considered. Blood cultures should be obtained, as well as mycobacteriologic cultures of sputum and blood and Interferon-Gamma Release Assays (IGRA), if tuberculosis is suspected. In granulomatous uveitis or choroiditis, tuberculin test might be positive; if tuberculosis infection is not documented, a hypersensitivity reaction to its antigens will have to be considered [7].
Adding anti-hepatitis C antibody and cryoglobulins if cryoglobulinemia is suspected, upper and lower gastrointestinal endoscopies if IBD is a possibility or a skin biopsy if suspicious lesions are present must be considered. The hypothesis of an intraocular lymphoma should always be excluded, especially in the elderly and in steroid-resistant uveitis; progressive and bilateral cellular infiltration of the vitreous, in contrast with few cells at anterior chamber, is suggestive [7].
Before starting treatment, infections, tumors and ophthalmologic diseases must be excluded, attending to different therapeutic approaches. If infection is identified, appropriate antimicrobial or antiviral therapy is mandatory before attempting immunosuppression for AIU. The identification of an underlying systemic disease is important not only to treat ocular inflammation but also to control life threatening complications of SARD.
Current treatments focus on immunosuppressive therapies to control acute inflammation and to ensure the maintenance of long-term remission. Corticosteroids are usually among the first chosen due to their effectiveness at controlling inflammation both in the short term and in the long term. However, a myriad of possible side effects as well as ocular sequelae may be observed [10].
The general therapeutic approach of AIU depends on ophthalmological semiology, including the topography and laterality of ocular lesions [7].
In general, AAU responds well to topical therapy with corticosteroids (betamethasone, dexamethasone or prednisolone, given every one to two hours until inflammation is under control and then tapered over the next four to six weeks), mydriatics and cycloplegic drugs (atropine or phenylephrine, one to two drops three to six times/day, until inflammation is completely controlled). Topical therapy needs to be given intensively and for an adequate period of time to prevent complications and reduce pain secondary to ciliary muscle spasm [3]. Failure to adequately treat an AAU may delay improvement and erroneously lead to the uveitis being considered as refractory. Patients should be followed weekly or biweekly during the phase of active inflammation. If more than three flares occur in a year or if two flares occur in less than three months, treatment with sulfasalazine is proposed to decrease the number of recurrences [3]. Subtenon corticosteroid injections with depot steroids are indicated in cases of non-compliance with topical treatment, no improvement after adequate topical therapy, extremely severe flares and when there is a tendency for recurrences or a chronic course after tapering topical treatment [3]. In cases of severe anterior uveitis, oral corticosteroids should be initiated [7]. Short courses of low-medium doses (less than 30 mg/day of prednisolone) are sometimes necessary, when all the treatments mentioned above are ineffective. Sulfasalazine or methotrexate could be an option for patients who fail to respond to steroids [3].
For intermediate or posterior uveitis, the laterality of the lesions determines the treatment: unilateral uveitis is treated with periocular injections of corticosteroids (administered as 40 mg triamcinolone through the posterior subtenon or orbital floor or alternatively as 40 mg methylprednisolone through the orbital floor), while bilateral lesions require systemic agents [7,22]. For vision threatening uveitis, chronic posterior uveitis, panuveitis or refractory CAU, oral prednisolone is started in a dose of 1 mg/kg/day for up to one month or until the disease is under control. For acute flares with sight threatening inflammatory disease, such as Behcet’s retinitis, treatment with high dose intravenous pulses of methylprednisolone may be helpful [23]. Progressive tapering should then follow (5-10 mg/week); maintenance therapy with low-dose prednisolone (2.5-10 mg/day) is often needed [11,23]. Steroid implants offer the benefit of sustained corticosteroid delivery to the eye while avoiding systemic complications of other therapies [22]. The use of intravitreous triamcinolone has been proven to be efficacious in patients that are non-compliant; it has been used in cases of BS, as well as in panuveitis. Vitreous corticosteroid implants using a small dose of fluocinolone acetonide are indicated when an effective, safer and longer action is attained [8]. In panuveitis, supportive therapy includes cycloplegic drugs, mydriatic agents, atropine in acute attacks and intermediate acting agents (homatropine) for maintaining pupillary dilatation [23].
Figure 1. Algorithm for the diagnosis of a suspected autoimmune uveitis [3,7,20].
Corticosteroid sparing agents are indicated when high dose steroids (more than 60 mg or less, based on the weight of individual dosed 1 mg/ kg) for more than one month or chronic doses greater than 7.5 mg/day are required for controlling ocular inflammation, as well as in cases of corticosteroid ineffectiveness, iatrogeny requiring discontinuation, uveitis refractory to appropriate tapering and in BS [3,22]. Patients are typically transitioned to steroid sparing therapy and the steroid is slowly tapered once the ocular inflammation is silent [22].
The choice of an immunosuppressive drug depends on disease and patient’s characteristics [7]. Treating chronic and relapsing AIU with these agents results in several side effects and the efficacy is far from excellent [5]. Steroid sparing agents include antimetabolites (methotrexate, azathioprine and mycophenolate mofetil (MMF)), calcineurin inhibitors (cyclosporine and tacrolimus), alkylating agents (cyclophosphamide and chlorambucil) and biologics (TNF-α inhibitors infliximab, adalimumab and etanercept; daclizumab, interferon α-2a and rituximab) [22].
Cyclosporine, azathioprine and methotrexate are the most frequently used immunosuppressiveagents to treat AIU. Nonetheless, only few clinical trials have been conducted to date, mainly with cyclosporine [3]. On the Systemic Immunosuppressive Therapy for Eye Diseases (SITE) Cohort Study [24], methotrexate, azathioprine, MMF, cyclosporine and cyclophosphamide all achieved at least 50% success at controlling ocular inflammation within one year [24]. Azathioprine (1-3 mg/kg/day) showed a moderate success on the same cohort.
Methotrexate (7.5-25 mg/week in conjunction with folic acid) is a widely used agent for ocular inflammation [22]. In the SITE Cohort Study it was shown to be moderately effective, mainly for anterior uveitis, with greatest success for the treatment of intermediate uveitis. MMF (500-1500 mg bid) has been shown to be effective in combination with steroids or another immunomodulatory treatment, as well as in monotherapy [22].
Randomized controlled trials (RCT) have shown cyclosporine effectiveness in ocular inflammation [22,25]. Initial treatment should combine 1 mg/kg/day prednisolone with 5 mg/kg/day cyclosporine divided into two doses up to a maximum of 10 mg/kg/day for refractory cases. Once steroids are withdrawn, cyclosporine can be slowly tapered down, 10% of the dose every month, to the minimum effective dose. Cyclosporine response can be seen in a few weeks in about 60% of cases, with a decrease in the number of flares. It has a dose-dependent effect, with exacerbations when it is decreased rapidly [3]. The use of tacrolimus (0.05 mg/kg/day) in uveitis is more limited but, in patients that become resistant or develop nephrotoxicity to cyclosporine, it was found to stabilize vision. Intravitreal sirolimus injection for the treatment of active AIU may be also an option [22]. Voclosporin has been recently used to treat non-infectious uveitis; limited data available indicate at least comparable results relative to current therapy, with a better safety profile [26].
Cyclophosphamide can be used for ocular inflammation (1-3 mg/ kg orally or intravenous pulse therapy 1gm/m2 body surface area every three to four weeks). In SITE cohort, patients taking cyclophosphamide achieved control of inflammation in 49% at six months and 76% within 12 months but also showed a trend for slightly increased cancer-related mortality [22,24]. Because of the risks associated with chlorambucil (0.1 mg/kg/day-15 mg/day), its use is generally restricted to severe sight threatening uveitis, such as in BS [22].
In cases of lack of efficacy or intolerance to corticosteroids plus immunosuppressive agents, biologic agents can be used. There is an ongoing argument as to whether biologics should remain as second-line treatment or be considered as first-line therapy in the management of certain uveitic entities, based on the results of the current uveitis literature regarding their efficacy and safety [8,11,27,28]. Infliximab is effective in AIU (recommendation C, evidence level 2b); an induction dose of 5 mg/kg at weeks 0, 4, and 6, administered in four to eight week intervals, depending on the clinical response, was the most frequent dosage regimen reported in a recent systematic review by Cordero-Coma et al. [22,25,28] Adalimumab (40 mg subcutaneous injection every one to two weeks) has also shown effectiveness in the treatment of AIU (recommendation C, evidence level 2b) while etanercept has been found to be ineffective (recommendation A, evidence level 1b) [22,28].
Interferon α-2a was successfully used in the treatment of severe uveitis associated with BS and prevented remissions. Interferon α-2b is an alternative agent. Rituximab has been used for ocular involvement in rheumatoid arthritis (RA), GPA and BS [22].
Ocular surgery is an option only in rare cases due to the high risk of relapse of uveitis and its complications [17]. The current treatment strategies are hampered by the paucity of RCT and few trials comparing efficacy of different agents. Much of the drug data is based on retrospective information. Patients need to be well aware of the risks and benefits of systemic treatment for ocular disease and whether they are willing to assume potentially life long risks versus the burden of further vision loss [22]. The better comprehension of the pathogenic mechanisms involved in the genesis of AIU will certainly lead to an optimized treatment. Therapeutic innovations for AIU are needed; the ideal therapy should improve the current disease process and prevent new events, in addition to being safe [5]. A systematic review on the effectiveness of immunosuppressants and biological therapies in the treatment of posterior AIU by the Uveitis Working Group from Spanish Society of Rheumatology was recently performed. In general, both immunosuppressive and biologic therapies are effective to treat posterior AIU, except for daclizumab in BS and etanercept in any type of uveitis. No superiority was inferred from this review [27].
Patients with AIU should be regularly followed by an expert ophthalmologist and referred to other physicians for studying and managing systemic manifestations [29]. The prognosis is generally good if patients receive prompt diagnosis and treatment, and serious complications (cataract, glaucoma, keratopathy, macular edema and permanent vision loss) develop only if untreated [17]. A close collaboration between ophthalmologists, internists and rheumatologists, in multidisciplinary consultations, is very important.
SS is a chronic multisystemic inflammatory disorder of unknown etiology that commonly affects young and middle-aged adults. Diagnosis is established when clinical and radiological findings are supported by the presence of non-caseating epithelioid cell granulomas and other granulomatous diseases are excluded. SS affects most often the lungs, lymph nodes, skin, heart, liver, muscles, and the eye.
Ocular involvement occurs in 10 to 50% of European and 50 to 90% of American patients with SS (11-83% during disease course and 1.5-12.4% at first presentation), corresponding to the most common extrapulmonary involvement. It is commoner in African Americans than in Caucasians. Isolated eye sarcoidosis can also occur [8,30-32]. Two peaks of incidence were reported for ocular sarcoidosis (OS), at the third and the fifth decades; sarcoid uveitis appears to occur more likely in African patients younger than 50 years old and in Caucasian patients older than 50 years old [21,30].
Any part of the eye can be affected but uveitis is the most common ocular manifestation of sarcoidosis (prevalence of about 30-70%) with anterior involvement often being self-limited, whereas posterior involvement can be chronic [30,31,33]. Any uveitic pattern can be found in sarcoidosis [2]. Anterior uveitis is more common than the posterior location and predominates in Blacks (70-75%); posterior uveitis is rarer but predominant in Caucasians (65-83%) [30,31]. Variable prevalences of SS as a cause of AIU have been reported: 2.4% in a cross-sectional study of 2619 uveitic patients (2.1% of the anterior uveitis, 1.5% of the intermediate uveitis, 2.5% of the posterior uveitis and 7.1% of the panuveitis cases) and 11.6% in another of 121 patients [5]. Ocular manifestations of sarcoidosis (unlike systemic) are not associated with polymorphisms in NOD2 [34].
OS consists on a granulomatous or non-granulomatous uveitis, anterior and/or posterior. In half of the cases, the presentation is an acute and selflimited granulomatous iridocyclitis. The chronic presentation is mostly seen in older patients with pulmonary fibrosis and quiescent systemic disease and is commonly manifested by an intermediate uveitis [8]. Although there is not a specific pattern for sarcoid uveitis, a granulomatous anterior uveitis, an intermediate bilateral uveitis, a bilateral panuveitis or granulomatous lesions at the choroid or at the optic nerve are suggestive of sarcoidosis, in the appropriate clinical and radiological setting [2].
AAU uveitis is characterized by “mutton-fat” keratic precipitates while CAU is associated with iris nodules (highly suggestive of sarcoidosis) and can lead to band keratopathy, glaucoma and cataract formation. In intermediate uveitis, the periphery of the retina may show “strings of pearls” or “snowballs”. The most common manifestation of posterior uveitis is RV, nearly always a retinal periphlebitis associated with segmental cuffing, sheathing and perivenous infiltrates, referred to as “candlewax drippings”, which may be subclinical and only identified with fluorescein angiography. The involvement of vessels is discontinuous and this appears clinically as skip lesions. It may be mild and associated with peripheral retinal and focal vitreal infiltrates, indistinguishable from idiopathic pars planitis. RV may be the presenting sign in patients who develop sarcoidosis many years later [16,33]. Chronic uveitis can lead to neurologic involvement and to cystoid macular edema that may be particularly refractory to conventional anti-inflammatory therapy [34,36].
Although many patients experience severe pain and photophobia, over a third of the patients do not have ocular symptoms. Therefore, it is recommended that all patients with sarcoidosis undergo annual ophthalmologic examination regardless of symptoms [33].
A first diagnostic approach (Table 2) should be followed to exclude infection. Because ocular disease may be the first manifestation of sarcoidosis, physicians should adapt a multidisciplinary approach to uveitis. The diagnosis of OS in patients with known sarcoidosis usually requires a specific examination by an ophthalmologist [33].
The International Workshop on Ocular Sarcoidosis consensus conference [36] has identified seven signs in the diagnosis of OS and pointed up the diagnostic strategy for its confirmation. This approach has been supported by a Japanese retrospective case-controlled study [37].
Testing for angiotensin-converting enzyme (ACE) and lysozyme in combination with chest imaging may identify the vast majority of patients with SS [21]. The gold standard for definitive diagnosis remains biopsy confirmation [33]. Because of the risk involved in ocular biopsy, it is rarely performed in patients with suspected OS; however, biopsy of conjunctival nodules is useful and less invasive [20,32]. Biopsy is usually performed from a more accessible site, such as the lungs, skin or a palpable lymph node. When there is no involvement of skin or surface lymph nodes, transbronchial lung biopsy (TBLB) has often been performed because of its effectiveness and relative safety, even in patients without pulmonary lesions. Takada et al. recently suggested a flow chart for survey of patients with suspected OS: if chest CT scan, serum ACE and bronchoalveolar lavage results are suggestive of SS (high clinical probability) or if chest CT scan is not suggestive of SS (low clinical probability), TBLB can be avoided [32]. Lung biopsies through mediastinoscopy appear to be informative but this invasive approach is indicated only for cases of differential diagnosis with lymphoma or in the presence of a severe uveitis needing systemic treatment [20]. In the absence of histologic confirmation of sarcoidosis, and even with normal chest CT scan, gallium scintigraphy and positron emission tomography scan may identify the presence of hyperfixating lesions suggestive of granulomas, allowing a presumptive diagnosis [20]. Indocyanine green angiography and optical coherence tomography have dramatically improved the understanding of choroidal granulomas and macular edema [38].
OS should always be treated but the optimal management has not been well defined. Corticosteroids are the mainstay of treatment, either topical or systemic, attending to the extent of ocular involvement. Steroiddependence is frequent and a state of controlled inflammation has been achieved and reported by a few authors using low-dose methotrexate, in the otherwise difficult-to-treat patients with panuveitis [23]. It has been proposed as first-line immunosuppressant in OS [7]. Combination immunosuppressive therapy has been discussed as a treatment approach for OS, with the most common combinations including methotrexate plus azathioprine and methotrexate plus leflunomide. Anti-TNF-α agents can be quite effective for chronic refractory OS, namely infliximab and adalimubab [33].
OS is potentially vision-threatening and approximately 20% of patients with uveitis will suffer some degree of visual loss [11]. Only the presence of cystoid macular edema was significantly associated with worst visual outcome [39].
SpA are a group of rheumatic diseases including ankylosing spondylitis (AS), psoriatic arthritis (PsA), reactive arthritis (ReA), enteropathic arthritis (EA) and undifferentiated arthritis (UA) [40]. Like HLA-B27, past or present uveitis, confirmed by an ophthalmologist, is a feature which is included in the Assessment of Spondyloarthritis International Society (ASAS) classification criteria for axial SpA [11]. Uveitis associated with SpA are related to the group of HLA-B27 positive uveitis [29].
AAU is the most common form of intraocular inflammation and about 50% of the cases are associated with HLA-B27 [40]. The prevalence of AAU in HLA-B27 population is about 1%, contrasting to 0.2% in the general population [41]. Affected patients are usually males (two to five times more often than women but lower ratios have been reported) and younger than those with HLA-B27 negative uveitis, usually in their forth decade of life [29,40]. However, once HLA-B27 uveitis is established, the risk of developing systemic disease may be similar for both genders [42]. It is typically associated with AS, occurring in 25 to 40% of patients, and less common in other forms of SpA (25% in PsA and ReA, 10-36% in EA) [11,40]. About 90% of AS patients with AAU are HLA-B27 positive [11]. Disease probability for SpA according to the presence of AAU is one of the highest for clinical parameters (likelihood ratio of 7.3) [11]. Besides HLA-B27 association, a microbial trigger (Klebsiella, etc.) has been suspected in AAU associated with SpA [9].
Clinically, AAU is almost always symptomatic and presents with unilateral and sudden redness, pain, photophobia, blurred vision, increased lacrimation and miosis, often self-limiting and recurrent, also affecting the contralateral eye [11]. Both eyes are affected in about 80% of cases of AAU, but usually not simultaneously, and females may be more prone to develop bilateral uveitis [42]. Comparing to CAU, AAU in SpA is less severe because inflammation is not constant, but it is generally more serious than other kinds of acute uveitis [29]. About 50% of recurrent unilateral AAU are associated with SpA [11]. Hypopyon may be occasionally seen [29]. Slit-lamp examination allows diagnosis and monitoring of response to treatment [11]. In SpA, rheumatologic symptoms usually precede uveitis, even though this can be the first sign of SpA in up to 11.4% of the cases [40]. Similarly, in EA, intestinal symptoms usually precede ocular ones, but these may constitute the initial manifestation and usually appear during the first years, being independent of bowel disease [29]. In PsA, uveitis is more often bilateral and prolonged [43]. Bilateral sacroileitis, HLA-DR13 and syndesmophytes are potential predictors of uveitis in PsA [44].
Posterior uveitis has been reported in SpA in up to 20% of the cases, being more severe mainly in those with IBD [11,16]. Conjunctivits has also been associated with AS [29]. Classically, ReA has been described as the triad of conjunctivitis, arthritis and urethritis, but the former is only present in 30 to 60% of cases, while keratitis and iridocyclitis are even rarer [29]. In PsA, conjunctivitis (20%) is also more common than the typical unilateral relapsing AAU (7-10%), which may rarely evolve to chronic uveitis [8,29]. In EA, episcleritis (up to 29%) and scleritis (18%) are more frequent than corneal inflammation, posterior uveitis (chorioretinitis in up to 10% of patients with IBD, but also RV), myositis, orbital pseudotumors and anterior uveitis. The last is more common in females and often presents with an insidious (but non silent) chronic course [29].
Topical glucocorticoid eye drops and mydriatics are sufficient in most cases but intraocular or oral glucocorticoids (up to 60 mg prednisone daily) may be necessary in more severe cases [11,43]. Nonsteroid antiinflammatory drugs have also shown some efficacy [41]. Sulfasalazine might be used to prevent attacks, while the role of TNF-α blockers for the treatment of severe AAU needs to be defined [11]. Infliximab and adalimumab seem to be more effective than etarnecept in decreasing the recurrence rate [41]. Treatment of triggering infection might alter the clinical course in ReA, whilst the treatment of skin manifestations in PsA does not alter the visual outcome [29].
Prognosis is usually favorable and symptoms often subside within weeks, but some reports suggest that blindness may reach 11% [11,41]. HLA-B27 positivity may be associated with a more severe course [16,44]. Complications of AAU include anterior and posterior synechiae, cataracts, cystoid macular edema and glaucoma. Macular edema is the main factor determining visual outcome and is present in 4 to 31% of cases [11,42].
BS is an immune-mediated, systemic, relapsing and chronic vasculitis of unclear origin, whose diagnosis requires recurrent oral ulceration and at least two of the following items: recurrent genital aphtosis, typical skin lesions, a positive pathergy test or characteristic eye involvement [45,46].
Ocular involvement in BS (ocular BS) occurs in about 50% of the patients, and up to 80% in some series [11,46]. It constitutes the initial symptom in about 10 to 20% of patients, and is often present in the first two years, appearing rarely after five years of disease duration [46]. Ocular BS is more prevalent in males (83-95% contrasting with 67-73% in females) and younger patients [8,11,46]. Eye involvement is one of the most devastating consequences of BS, along with central nervous system involvement [46,47].
Ocular BS usually presents with a chronic, non-granulomatous and relapsing uveitis, with necrotizing obliterative RV, often bilateral (78-95%) and affecting anterior and (mainly) posterior uveal tracts [11,46-48]. In fact, panuveitis is estimated to occur in between 40% to more than 60% of those with ocular involvement and is probably more common than isolated posterior uveitis [47,49]. Initially, unilateral anterior involvement may occur, but bilateral panuveitis often takes place, affecting mostly and more severely the posterior segment of the eye [46]. Posterior uveitis can present with no symptoms or simply with loss of vision with no ocular pain [49]. Associations have been reported with NOD2 and IL-15 polymorphisms in BS uveitis, but an infectious trigger is also suspected [9]. RV is characterized by an occlusive and necrotizing vasculitis at the posterior pole and the involvement of small and medium-sized vessels is a pathognomonic finding, even though veins are mostly affected [46]. Posterior eye involvement can also manifest as cell infiltration of the vitreous body or edema affecting the retina, macula or optic disc [46]. Acute periphlebitis and thromboangiitis obliterans, with possible massive retinal and vitreous bleeding, may result from inflammation of retinal vessels [46]. Retinal edema is a consequence of recurrent inflammatory activity, but visual loss often follows retinal atrophy [46]. Neovascularization, retinal detachment, cataracts and secondary glaucoma may complicate the clinical course of posterior ocular BS [46]. Fluorescein angiography is helpful for RV diagnosis, even when fundoscopy is normal [8].
BS was originally described as a triad of hypopyon-iritis, recurrent oral and genital ulcerations [46]. Hypopyon uveitis is seen in about 20 to 30% and occurs as a solitary finding in about 10% of patients with ocular BS, being invariably associated with severe retinal disease [11,46]. Nowadays, hypopyon iritis is less common but one should be aware that optic atrophy is expected in about half of the patients; other complications include anterior and posterior synechiae, secondary glaucoma, cataracts and macular edema [46]. Red eye and photophobia result from anterior uveitis [11]. Less common lesions of the anterior eye include conjunctivitis, sicca syndrome, episcleritis, scleritis, keratitis, lid lesions and extraocular muscle involvement [11,46].
Topical treatment with corticosteroids, mydriatic and cycloplegic agents is usually restricted to ocular BS with mild to moderate anterior inflammation [46,50]. Hypopyon uveitis responds to high doses of corticosteroids within days, but relapses may occur when doses are lowered [50]. Azathioprine, with or without systemic corticosteroids, is often recommended as first line treatment for ocular involvement in BS, especially with severe uveitis involving the retina or associated with visual loss [11]. The addition of cyclosporine, interferon-α or infliximab can be considered in refractory cases [11]. Cyclophosphamide and possibly thalidomide may also prevent ocular manifestations [50]. Despite the use of immunosuppressive drugs, loss of useful vision is seen in up to 74% of affected eyes within five to ten years of disease [46].
Ocular BS is more severe in males, younger patients and those with hypopyon uveitis, synechiae and retinal scars [11]. Regardless of immunosuppressive treatment, significant visual impairment occurs in 25 to 50% of patients [16].
VKH is a rare multisystemic disease that affects tissues containing melanocytes, such as the eyes, central nervous system, inner ear and skin. It occurs more frequently among people with darker skin pigmentation, including Asians, Indians and Latin Americans. In Japan, 8% of all cases of uveitis are caused by VKH. Females are affected more often, as well as people in their second to fifth decades of life [52,53]. In a cross-sectional study of an uveitis cohort (n=2619), VKH was present in 11 patients (0.42%), accounting for 1.8% of the posterior uveitis and 3% of the panuveitis cases [24].
The cause of VKH remains unknown but it is thought to result from a T-cell-mediated autoimmune process directed against melanocytes. HLA DRB1*0405 is the most common allele found in association with VKH [34,52,53].
The classic clinical course can be divided into three stages: stage 1 or prodromal stage includes headache, fever, and photophobia and may mimic a viral infection; stage 2 or ophthalmic stage includes severe bilateral diffuse granulomatous uveitis, bilateral neuro-sensorial dysacusis, meningitis and cutaneous involvement with poliosis, vitiligo, and alopecia; and stage 3 or convalescence stage can last for weeks or months [8,53] .
Ocular findings are characterized by bilateral posterior uveitis with retinal edema, hyperemia or edema of the optic disc, serous retinal detachments, iridocyclitis and increased intraocular pressure. The anterior ocular chamber may also be involved with formation of “muttonfat” keratic precipitates and iris nodules. Other typical ocular findings are “sunset glow” fundus, Dalen-Fuchs nodules and migration or accumulation of the pigmented epithelium of the retina. The most common symptom of VKH uveitis is bilateral blurred vision, which is present in 70% of the cases. Other symptoms include ocular pain, photophobia, conjunctival hyperemia, loss of visual acuity and blindness. Although rare, unilateral ocular involvement can be observed [52].
Repeated outbreaks of ocular inflammation may occur with multiple ophthalmological complications such as repeated retinal detachment, glaucoma, formation of subretinal neovascular membranes, subretinal fibrosis and, most commonly, cataracts [52].
The suspicion of VKH is essentially clinical but certain supplementary tests can indicate the presence of inflammatory activity and help to investigate differential diagnosis and identify concurrent diseases (Table 2). The most common ocular imaging exams are angiography with fluorescein or indocyanine green, the second of which is very sensitive for detecting subclinical injuries to the choroid [53]. Criteria for the diagnosis of VKH were revised in 2001 [54].
VKH should promptly be treated with systemic high-dose corticosteroids, generally prescribed at 1-2 mg/kg/day of oral prednisolone. For more severe cases, pulse therapy is given with 1 g/ day of methylprednisolone for three to five days [23,52,53]. For patients who do not respond well to corticosteroids or develop side effects, non-steroid immunomodulatory treatment is recommended and it has been considered “mandatory” in VKH. Options include methotrexate, tacrolimus, cyclosporine, MMF, azathioprine, cyclophosphamide and chlorambucil [52,53]. Cyclosporine has been used most broadly and effectively to control inflammation in VKH patients, limited to 5 mg/kg/ day to minimize toxicity [53]. Patients who present with aggressive forms of VKH may benefit from triple immunosuppression with prednisolone, azathioprine and cyclosporine. In cases of corticosteroid-resistance that also fail to respond to immunosuppressants, intravenous immunoglobulin may be a viable treatment option [54].
Biologic agents are an alternative therapy currently being used to treat VKH, including anti-TNF-α agents (infliximab and adalimumab) and anti-vascular endothelial growth factor agents (bevacizumab and ranibizumab) [55]. The treatment with an intravitreal single injection of bevacizumab in patients with neovascular membranes secondary to intraocular inflammation is being studied and was demonstrated to be effective [8].
Visual prognosis is highly variable, but generally favorable. It has been found that the three most important prognostic factors are good visual acuity one month after starting treatment, early high-dose corticosteroids and younger age at disease onset [52].
In practice, rheumatoid vasculitis is considered when clinical manifestations of vasculitis with intraocular inflammation, unexplained by other conditions, occur in a patient with an established diagnosis of RA. Approximately 16% of patients with RA have ophthalmic manifestations, including scleritis and peripheral ulcerative keratitis (PUK). In a patient with RA, necrotizing scleritis or PUK usually herald systemic vasculitis and, if left untreated, are associated not only with visual loss but also with a mortality rate of up to 30% [16,55]. Angiographic evidence of RV has been reported in 18% of RA patients without clinical signs. RV and geographic choroiditis have also been reported as rare manifestations of RA [16].
SLE can involve any region of the eye, with keratoconjunctivitis sicca and RV being the most common manifestations [11]. Different prevalences of SLE in patients with uveitis have been reported, ranging from 0.1 to 4.8% [56]. The prevalence of SLE-RV is estimated to be about 10%, rising to 29% when fluorescein angiography is used for diagnosis, as there are many asymptomatic cases [2]. SLE-RV is a potentially blinding condition and an important marker of disease activity, being associated with neurolupus [16]. Therefore, close monitoring and aggressive treatment of these patients is critical [2]. Anti-phospholipid antibodies and lupus anticoagulant play a pathophysiological role in SLE-RV by increasing the risk of vasoocclusive retinopathy and large vessel occlusions; the deposition of immune complexes might also be involved [2,16]. The classic lesions in SLE-RV are “cotton-wool” spots, which represent focal areas of ischemia and are believed to result from occlusion of the small retinal arterioles by infiltrating inflammatory cells [2]. Other findings include intra-retinal hemorrhages, hard exudates, microaneurysms and structural vascular changes. Severe ischemia may result in retinal neovascularization, which can be complicated by sight threatening vitreous hemorrhage, tractional retinal detachment, and neovascular glaucoma [16]. Uveitis, though rare, may also occur in the absence of retinal involvement. Cases of anterior uveitis in SLE have been described but are exceptional [2]. In SLEassociated anterior uveitis, topical corticosteroids may be sufficient [57].
Ocular manifestations of systemic vasculitides may be observed in many forms, including conjunctivitis, episcleritis, scleritis, PUK and uveitis. Prompt recognition and association of ocular symptoms with a diagnosis of systemic vasculitis is critical to prevent permanent visual sequelae [16]. It is estimated that about 20% of patients with polyarteritis nodosa have ocular involvement (being PUK the most common manifestation). Multifocal RV is also frequent and it affects mainly arterioles, due to the deposition of immune complexes on vessel walls. Fibrinous anterior uveitis may occur but it is not a usual presentation [2]. In GPA, ocular or orbital involvement occurs in 28 to 87% of the patients, being an inaugural manifestation in 16% [2]. Although the most common manifestation is orbital granuloma, GPA can affect any ocular or periocular tissue [16,55]. Granulomatous anterior uveitis is the third most common ocular manifestation in GPA (10-20%) [2]. Retinal involvement is a relatively uncommon ophthalmic manifestation (5-12%), with retinal hemorrhages in the peripheral or posterior retina being the most frequent finding.
TINU is a rare specific form of intraocular inflammation combined with kidney disease. Its etiology is unknown. It is more frequent in children. A diagnosis of TINU generally accounts for 1 to 2% of all patients attending specialized uveitis centers [58]. TINU is a common cause of uveitis among patients who present with bilateral, non-granulomatous anterior uveitis of sudden onset (9% of the causes) and this subtype accounts for 80% of the uveitis in TINU. Ocular symptoms are often the forerunners of renal symptoms up to 14 months before in 65% of cases. Although the activity of eye disease can persist for many months, the outcome is usually favorable [3,7,8,58]. As the inflammation is mainly anterior, local treatment in the form of drops or injections has an obvious appeal. Still, it has to be borne in mind that renal disease may coexist and may benefit from systemic treatment. Ocular disease requires low-dose topical treatment regimen for months [11,58].
The clinical management of AIU is a challenging process. It is desirable that, in the future, an increasing awareness of AIU will surpass its under-recognition as an important cause of blindness. SS, SpA, BS and VKH are SARD in which the prevalence and characteristics of AIU are reasonably defined. In other SARD, including some cited in this review, these features are relatively unknown, as it happens with the link between ocular inflammation and manifestations in other organs. It is probable that each step taken towards a better clinical management of AIU in each disease will enhance the knowledge of AIU associated with other SARD, but it must not be forgotten that each one has its particularities. A multidisciplinary approach will certainly continue to be essential to the optimal management of AIU; uveitis is a perfect example of how specialists from different fields should collaborate to obtain solid diagnoses in complex conditions [17]. Even though several diagnostic and treatment algorithms will probably appear and guide clinical management, one should never dismiss an individualized approach of AIU. It is expected that further insight into the pathogenesis of AIU will allow the definition of new possible therapeutical targets, the development of new drugs or just the application of those agents which are already used in other SARD and have not been tried in AIU yet. Moreover, RCT are needed not only to meet these purposes but also to allow a better definition of doses and other pharmacokinetic parameters for corticosteroids, immunosuppressants and biologic agents that are already used in common clinical practice. With a better comprehension of specific mechanisms, we cannot discard the possibility that some drugs that are actually considered only for refractory cases will be considered as first line therapies. It is also possible that some immunosuppressive agents that were used in the past will no longer be considered to treat AIU due to their low efficacy or toxicity profile.
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Download Provisional PDF Here
Article Type: Review Article
Citation: Chora I, Borges T, Dias C, Ourique C, Vilaça J, et al. (2015) Clinical Management of Uveitis in Systemic Rheumatic Autoimmune Diseases in Adults. Autoimmun Infec Dis 1(1): doi http://dx.doi.org/10.16966/2470-1025.104
Copyright: © 2015 Chora I, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access