
Figure 1: Renal parenchymal infiltration with B-lymphocytes.
Hassan N Al Dhneem Reem Al-Thwainy Mohammed A Al-Yahya Ibrahiem Saeed Abdul-Rahman*
Department of Internal Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia*Corresponding author: Ibrahiem Saeed Abdul-Rahman, Department of Internal Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, E-mail: isaeed@iau.edu.sa
Hematological malignancies can affect the kidneys in different ways. There may be direct invasion by the tumor cells, or the malignancy may act indirectly via immunologically mediated mechanisms. Primary Renal Lymphoma (PRL) without evidence of extrarenal spread has also been reported. The existence of this entity, however, has been questioned, because the kidneys do not normally contain lymphoid tissue. Renal involvement is rare in leukemias, and in some leukemias, renal dysfunction is usually found during the blastic crisis. Renal infiltration of leukemic cells has been recognized in some patients. In addition, some types of hematological neoplasia are associated with severe hypercalcemia that can lead to nephrocalcinosis. Renal involvement is one of the major manifestations of Multiple Myeloma (MM) and is an important cause of renal failure in the elderly. Renal failure occurs in more than 50% of MM patients and is usually caused by the so-called myeloma kidney. Tumor Lysis Syndrome (TLS) is an oncological emergency characterized by a combination of metabolic disorders observed at the start of treatment of hematological malignancies. TLS may also be associated with the advancement of aggressive lymphomas and leukemias. The syndrome is frequently associated with renal dysfunction. Bone marrow transplantation for treatment of selected hematological neoplasms can be complicated by renal failure resulting from a variety of causes. Early renal injury most often results from infection and its subsequent treatment. Late renal injury after bone marrow transplantation, characterized by a syndrome similar to the hemolytic uremic syndrome, is called Bone Marrow Transplant (BMT) nephropathy. This article reviews the clinical and pathological features of renal injury in hematological malignancies.
Leukemia; Lymphoma; Multiple myeloma; Tumor lysis syndrome; Acute renal failure; Plasma exchange; Hemodialysis
CLL is a clonal lymphoproliferative disorder with over 5 x 109 /L peripheral B-lymphocytes is to this day considered to be the most common adult leukemia and is usually diagnosed at a relatively older age (>65), hence it could be considered a disease of the elderly. Once diagnosed, it is monitored conservatively until it becomes symptomatic [1]. One of its clinical manifestations is renal involvement (Figure 1-4), although rare and poorly described which can occur through various mechanisms such as leukemic infiltration, glomerular disease such as membranoproliferative glomerulonephritis (MPGN) and medication side effects (Figures 5&6) [1]. Kidney involvement at diagnosis or during conservative follow up has been found to be an indicator of adverse outcomes.
Leukemic infiltration in CLL: Regardless of the type of leukemia, Acute Kidney Injury (AKI) is one of the most common complications of hematological malignancies. In one study conducted on patients with hematological malignancies admitted to the Intensive Care Unit (ICU) the mortality due to AKI was estimated to be 72% [2]. The causes for AKI can be divided into prerenal, intrarenal and postrenal. Of the intrarenal. Leukemic infiltration can cause microvascular compression and intrarenal obstruction, or it can lead to chronic inflammation and cytokine release. It usually follows an asymptomatic course, however, when clinical manifestations do occur renal insufficiency is the presentation. Interestingly, there appears to be no association between the level/extent of infiltration and severity of renal injury [3-5]. Figure 1-4 illustrate leukemic cells infiltrating the renal parenchyma.
Figure 1: Renal parenchymal infiltration with B-lymphocytes.

Figure 2: Glomeruli with focal segmental necrosis and lymphoid cell infiltration around glomeruli.

Figure 3: Glomeruli without pathological abnormalities visualized by LM with lymphocyte-like cell infiltration in the interstitial tissue.
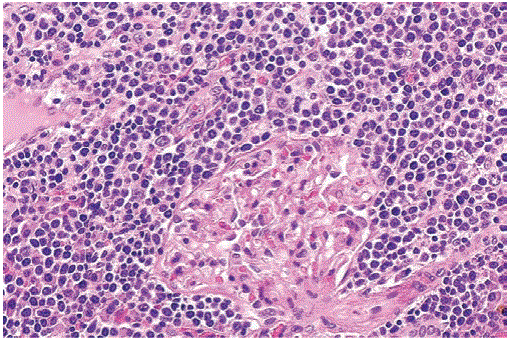
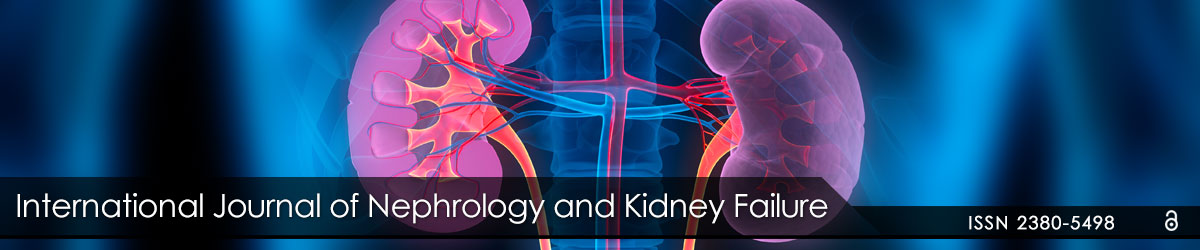
Figure 4: The tumor nodules are composed of small lymphocytes with rounded nuclei resembling chronic lymphocytic leukemia/small lymphocytic lymphoma. However, mantle cell lymphoma (MCL) lacks proliferation centers (growth centers) and prolymphocytes and paraimmunoblasts are absent. MCL is a mature B-cell neoplasm that expresses B-cell markers CD19, CD20, CD22, and CD79a. It has a tendency to express lambda light chains more frequently than kappa. Most cases of MCL carry the characteristic translocation t (11;14) (q13;q32) which results in juxtaposition of CCND1 gene next to IGH enhancer resulting in overexpression of cyclin D1.
Glomerular disease in CLL: Membranoproliferative Glomerulonephritis (MPGN) is the most common glomerular pathology accompanying CLL. Its clinical manifestation might vary from isolated hematuria/proteinuria to full blown nephrotic or nephritic syndrome. Its pathogenesis is postulated to be related to immune-complex deposition, cryoglobulin, light chain and monoclonal IgG as demonstrated on Immunofluorescent staining of renal biopsy (Figure 5) as well as electron microscopy (Figure 6) [6]. In addition, AL Amyloidosis, which are amyloid fibrils derived from monoclonal IgG light chains can also lead to systemic amyloidosis with kidney involvement, and interestingly the type of light chain involved is different than the usual ones implicated in plasma cell dyscrasia (lambda). Of the cases reported it was found that kappa chain was involved. Moreover, it was also found that they are associated with a more favorable prognosis than those implicated in plasma cell dyscrasia. MPGN associated CLL was also found to have significant improvement in renal abnormalities after receiving CLL directed therapy cyclophosphamide, prednisolone, and rituximab [7].

Figure 5: MPGN on immunofluorescence showing granular pattern with IgG and kappa light chain deposit.

Figure 6: Electron microscopy of MPGN showing subendothelial deposit (white arrow) and mesangial deposit (red arrow).
Treatment-related toxicities: The standard regimen of CLL includes fludarabine in combination with cyclophosphamide and rituximab. However, there has been a shift in using novel agents that are more specific to dysregulated pathways in the pathogenesis of CLL due to the toxicities and increased rates of infection associated with the old regimen. The novel agents include the monoclonal antibody Obinutuzumab, the Bruton’s tyrosine kinase inhibitors ibrutinib and acalabrutinib, the phosphatidylinositol 3-kinase inhibitor idelalisib and the BCL-2 inhibitor venetoclax [5]. Because these agents are relatively new and not widely used, the reported rate of renal toxicity might be undervalued, however, of the noteworthy adverse effects that were reported in the literature were related to electrolyte disturbances such as hypo/hypernatremia, hypo/hyperkalemia, and hypo/ hypercalcemia, in addition to AKI [5].
ALL is the most common type of cancer in the pediatric population, its usual presentation at diagnosis is organomegaly (liver, spleen) and signs of anemia with renal manifestation occurring at the end of the disease such as nephromegaly and high blood pressure (HBP). The incidence of nephromegaly in leukemic patients ranges between 2-24% and the pathology behind it is said to be due to leukemic cell infiltration and is frequently asymptomatic. However, when clinical manifestations due to kidney involvement do occur, they present as AKI, hematuria or HBP. There are few cases reported about kidney involvement in ALL, primarily because it presents very late in the disease and is not as common as hepatosplenomegaly and other signs of leukemia. Nonetheless, from the cases reported, the high blood pressure could be due to the kidney involvement itself or as a side effect of the medications used such as steroids [8].
CML is a type of the myeloproliferative disorders family that results from unchecked neoplastic proliferation of pluripotent stem cells that arrest in one of the myeloid Tri-lineage (polymorphonuclear, erythroid, megakaryocytic). CML is distinguished from the rest of the myeloproliferative disorders by the Philadelphia chromosome, a chromosome that’s created by the fusion of the ABL-BCR gene due to the T (9:22) translocation. It classically manifests with symptoms of bone marrow suppression such as anemia and easy bruising, massive splenomegaly, and hyperuricemia. Kidney involvement is infrequently seen and usually occurs late in the disease, specifically the blast phase. In one of the cases reported in the literature, the patient was diagnosed with CML for 6 years and presented with a suspected kidney tumor that was eventually diagnosed as leukemic cell infiltration of the kidney in the blast phase after doing a kidney biopsy (Figure 7) [9].

Figure 7: Renal biopsy showing immature myeloid cells infiltrating the renal parenchyma.
MDS results from maturation defect, i.e., dyspoiesis, leading to ineffective hematopoiesis and causing pancytopenia. The main feared complication of MDS is leukemic transformation to acute myeloid leukemia. Kidney involvement in MDS is extremely rare and is defined on a broad spectrum of kidney diseases proven by biopsy which include acute Tubulointerstitial Nephritis (TIN) and autoimmune Glomerulonephritis (GN) in particular, membranous nephropathy. Furthermore, it is speculated that the association between TIN and GN is due to MDS-linked autoimmune dysregulation [10].
Lymphomas are malignancies arising from the lymphoid system; this enables the disease to manifest at any site where lymphoid tissue is present [11]. The WHO classification of tumors of hemopoietic and lymphoid tissues primarily distinguishes Hodgkin Lymphoma (HL) from Non-Hodgkin Lymphoma (NHL). HL is subclassified as Nodular Lymphocyte-Predominant HL (NLPHL), and Classic Hodgkin Lymphoma (CHL) that is further subdivided into nodular sclerosing, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted. Treatment of HL is predominantly by chemotherapy, compromised of doxorubicin, bleomycin, vinblastine, and dacarbazine, followed by radiotherapy. In the late stages of the disease, the regimen can be escalated to include etoposide phosphate, cyclophosphamide, procarbazine, and prednisone. If the disease remains resistant or relapsed, then a more intense therapy consisting of the previously mentioned regimen followed by high-dose therapy with carmustine, etoposide, cytarabine, and melphalan. Regarding NHL, depending on the stage, it can be treated by radiotherapy alone or chemoimmunotherapy incorporating cyclophosphamide, doxorubicin, vincristine, prednisolone, and an anti-CD20 monoclonal antibody (either rituximab or Obinutuzumab) [12].
Lymphoma and its treatment can lead to devastating morbidities, especially when advanced or with an intense treatment regimen. One of the organs that are commonly affected is the kidney, where its involvement can be classified as Primary Renal Lymphoma (PRL), Secondary Renal Lymphoma (SRL), or due to treatment toxicities [13]. An autopsy-based study published by Flan et al. demonstrated that NHL accounts for 30-60% of renal involvement cases in lymphoma [14]. Those with increased risk were patients with immunocompromised status and uncontrolled Epstein-Barr virus proliferation, organ transplantation, HIV infection, ataxia-telangiectasia, chronic inflammatory diseases, or undergoing chemotherapy [13].
PRL is a rare phenomenon, compromising only 0.7% of extranodal lymphomas, in which lymphoma involves the kidneys alone without evidence of disease elsewhere [13]. The pathogenesis of primary renal lymphoma is poorly understood as, anatomically, there is no lymphoid tissue in the kidney for the lymphoma to originate from. Three main hypotheses for the pathogenesis of PRL have been suggested. First, Puente Duanay PN, et al. hypothesized that an inflammatory process recruits lymphoid cells that become the beginning of the oncogenic process and ultimately leads to PRL [15]. Second, Salem Y, et al. attributed the renal capsule as the source due to its abundant lymphoid tissue, which may lead to the lymphoma genesis there and subsequently propagate to the parenchyma, presenting as PRL [16]. Finally, another explanation by Betta PG, et al. viewed the perirenal lymphoid tissue as the origin of the carcinogenesis process [17]. PRL tends to present in middle and advanced-age patients and is more often bilateral; on the other hand, children usually are affected unilaterally [18].
SRL is when the kidneys are involved in the presence of widespread nodal or extranodal lymphoma. The involvement of the kidneys varies in systemic lymphomas. Six presentations have been reported in the literature: lymphomatous infiltration, minimal change nephrotic syndrome (MCNS), monoclonal Ig deposition, amyloidosis, immunotactoid glomerulopathy, and membranous glomerulopathy [19].
Lymphocytic infiltration is the commonest presentation, accounting in 34% of SRL and 13% of HL patients in the largest case series study on the topic. Of the 34%, only 14% had been diagnosed with Lymphocytic infiltration of the kidneys before death [14]. This is due to the asymptomatic presentation of such a condition. Some manifestations suggesting lymphocytic infiltration are kidney failure, bilateral kidney enlargement, and sub-nephrotic range proteinuria. Pathological findings include normal tubules and glomeruli, with infiltration of monomorphic lymphocytes [20] (Figure 8). The infiltration pattern can be diffuse, multinodular, or a single large tumor [21].

Figure 8: Mononuclear cell infiltration in the peritubular capillaries.
MCNS is seen in 0.4% of all HL and accounts for 40% of HL glomerulopathies. It usually presents early in the course of the disease, preceding the lymphoma diagnosis in a third of the patients [22]. It is hypothesized that the condition is mediated by an unidentified soluble paraneoplastic permeability factor, leading to the loss of selective capillary permeability and allowing albumin and other negatively charged molecules to cross the glomerular barrier [23].
The pathophysiology of monoclonal Ig deposition is similar to that in multiple myeloma, where in lymphoplasmacytic lymphomas, light chains, heavy chains, or both are deposited in the glomeruli leading to proteinuria and possibly kidney failure [24].
Amyloidosis is a rare complication of Hodgkin lymphomas in advanced stages or patients with prominent inflammatory states. It can present as heavy proteinuria, anasarca, and filtration impairment from hypoalbuminemia-induced volume contraction and hypotension [23].
Immunotactoid Glomerulopathy (ITG) is a complication of lymphoplasmacytic disorders with a poor prognosis, where 50% of the patients develop end-stage renal disease by five years [25]. There are monomorphic microtubules larger than amyloid fibrils on kidney biopsy, staining negative for Congo red, with IgG and complement staining on immunofluorescence [26].
Membranous Glomerulopathy (MGN) is characterized by glomerular basement membrane antibody deposition, but the provoking antigen has yet to be determined. Treatment of lymphoma related MGN focuses on the underlying malignancy. The response has been mixed in case reports; several patients recovered kidney function with systemic chemotherapy, but no long-term follow-up was available [27].
Renal involvement, one of the major manifestations of Multiple Myeloma (MM), is an important cause of renal failure in the elderly. Renal failure occurs in more than 50% of MM patients [28,29] and is usually caused by a myeloma kidney (discussed below). The degree of renal failure is generally moderate and reversible in up to 50% of patients, particularly when it is related to such precipitating factors as hypercalcemia [30]. When renal failure is present, MM has a poor prognosis [29,30]. Despite its frequency and poor prognostic significance, few reports deal with the outcome of patients with MM and impaired renal function. Renal diseases associated with MM include the following: 1. Light-Chain Deposition Disease (LCDD) 2. Myeloma kidney 3. Acute renal failure 4. Renal tubular dysfunction 5. Hypercalcemia and radiocontrast agents’ effect 6. Plasma cell infiltration (Figure 9).

Figure 9: Leukemic infiltration in a patient with chronic lymphocytic leukemia.
Most of the renal diseases in MM are related to overproduction of monoclonal immunoglobulin light chains. The risk for renal dysfunction increases with the amount of light chains excreted. The risk increases from 7% in patients who excrete <0.005 g/day to 39% in those who excrete >2 g/day [30-32]. Serum or urinary protein electrophoresis is no longer recommended, because of its limited sensitivity [33]. In contrast to amyloidosis, the deposits in approximately 80% of patients with LCDD are composed of kappa rather than lambda light chains. The deposits are also granular in nature, do not form fibrils or beta pleated sheets, do not bind Congo red stain or thioflavin T, and are not associated with amyloid P protein. In amyloidosis the fibrils are usually derived from the variable region of the light chains, whereas in LCDD it is usually the constant region of the immunoglobulin light chain that is deposited [33,34]. This may explain the far brighter immunofluorescent staining for light chains found in LCDD than of those found in amyloidosis. The pathogenesis of the glomerulosclerosis in LCDD is not entirely clear, but mesangial cells from patients with LCDD produce transforming growth factor-β, which acts as an autacoid and promotes these cells to produce matrix proteins, such as type IV collagen, laminin, and fibronectin. Patients with LCDD are generally >45 years of age. Many such patients develop frank myeloma, and others clearly have lymphoplasmacytic B cell disease such as lymphoma or Waldenström macroglobulinemia. Even in such patients without overt plasma cell dyscrasia, it is the excessive production of abnormal monoclonal light chains that produce the disease [34]. As with amyloidosis, the clinical features vary with the location and extent of organ deposition of the monoclonal protein. Patients typically have cardiac, neural, hepatic, and renal involvement; but other organs such as the skin, spleen, thyroid, adrenal, and gastrointestinal tract may be involved [32,33]. Patients with renal involvement usually have significant glomerular involvement (Figures 10&11), and thus present with proteinuria. The nephrotic syndrome appears in as many as one-half of these patients, often accompanied by hypertension and renal insufficiency. Some patients have greater tubulointerstitial involvement and less proteinuria, along with renal insufficiency [31]. The prognosis for patients with LCDD is uncertain but appears to be better than that for amyloidosis. As with amyloidosis, death is often attributed to cardiac disease and heart failure or infectious complications [35]. In a large series of 63 patients, 65% of the patients developed myeloma [35]. Of the total 63 patients, 36 developed uremia and 37 died. Predictors of worse renal outcome included increased age and elevated serum creatinine at presentation. Predictors of worse patient survival included increased age, occurrence of frank myeloma, and extrarenal deposition of light chains. Although there are few data on dialysis and transplantation in LCDD, patients appear to fare as well as those with amyloidosis. Recurrences within the renal transplant have been reported. One trial of seven patients with LCDD who received renal transplants found recurrences in five of seven within a mean time of <1 year [36,37]. Thus, suppression of the abnormal paraprotein-producing cell clone is crucial prior to renal transplantation.

Figure 10: Plasma cell infiltration of the kidney in a patient with multiple myeloma. Jones’ silver stain, 400x.

Figure 11: Plasma cell infiltration of the kidney in a patient with multiple myeloma.
Myeloma kidney should be suspected in older patients who present with unexplained acute or subacute renal failure, normal-sized kidneys on ultrasonography evaluation, bland urinary sediment, negative or trace-positive Albustix test, and a markedly positive sulfosalicylic acid test. Myeloma kidney results when light chains with a predilection for cast formation are delivered to the distal tubule at a critical concentration. Light chains are filtered freely, absorbed by endocytotic receptors, and catabolized in proximal tubular cells [38]. The concentration of light chains reaching the distal tubule depends on the filtrate concentration, and on the capacity of the proximal tubule cells to absorb and catabolize them. Any reduction in the glomerular filtration rate or proximal tubular damage increases distal tubular delivery [28,29,38]. At a critical concentration, light chains aggregate and co-precipitate with Tamm-Horsfall proteins to form casts that obstruct tubular flow [39] (Figure 12). Hemodialysis has been described as a treatment for this disorder. Other factors that affect cast formation include the following [14,15-17]. 1. Distal nephron sodium, chloride, and calcium concentrations 2. Tubular flow rate 3. Presence of furosemide or radiocontrast agents [38] 4. Acidity of the urine 5. Concentration of the carbohydrate content of Tamm Horsfall glycoprotein [37-39] the diagnosis is confirmed by demonstrating monoclonal immunoglobulins in the serum or urine and typical intraluminal cast formation observed in a kidney biopsy specimen.
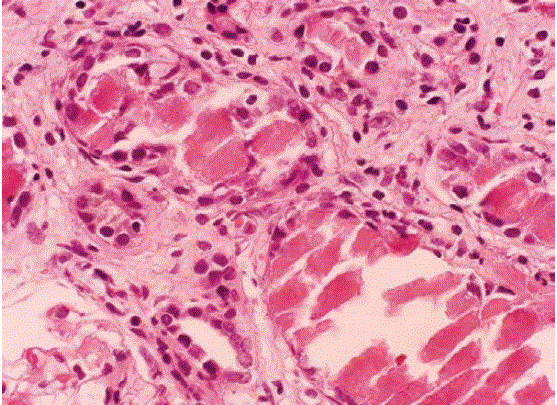
Figure 12: Myeloma cast nephropathy. Close-up of intratubular refractile casts with surrounding syncytial giant cell reaction. Note the chronic tubulointerstitial nephritis and fibrosis, characteristic of myeloma cast nephropathy. (PAS, 400×).
In the proximal tubules, there is accumulation of light chains that are resistant to proteolytic degradation. They form intracellular crystals and cause tubular dysfunction. This latter can present as Fanconi’s syndrome, with proximal tubular acidosis, aminoaciduria, hypouricemia, and phosphate wasting that leads to osteomalacia [17]. Proximal cell damage also results in decreased proximal clearance of light chains, thereby promoting cast formation distally [18]. About 15% of patients with MM are hypercalcemic, with a serum calcium concentration >2.75 mmol/L at diagnosis. Hypercalcemia contributes to renal failure by causing vasoconstriction, inducing hypovolemia through nephrogenic diabetes insipidus, and intratubular calcium deposition [19]. In patients with MM, the incidence of ARF caused by the use of radiocontrast agents ranges from 0.6% to 1.25% [20]. Contrast medium is thought to bind to intratubular proteins, causing them to precipitate and obstruct tubular flow. To prevent contrast nephrotoxicity, patients with MM must be well hydrated, and use of N-acetylcysteine should be considered [20,21]. Plasma cell infiltration of the kidney is seen in MM but is rarely severe enough to cause renal dysfunction [23].
Although some patients with MM do not require treatment, oncology referral is recommended in all cases. Patients with smoldering (asymptomatic) MM should not undergo treatment, as current research shows that starting active therapy for people with no symptoms does not improve survival [24]. Careful follow-up, however, is recommended. A systematic review by He, et al demonstrated a reduction in vertebral compression and time to progression with early systemic treatment for asymptomatic patients, but their study also revealed an increase in acute leukemia in the early-treatment group [25]. An important study that evaluated the risk of disease progression in asymptomatic subjects with MM showed that the patients did not benefit from early treatment, and delayed treatment did not affect the efficacy of therapy in terms of survival [26].
Initial treatment of MM depends on the patient’s age and co-morbidities. In recent years, high-dose chemotherapy with hematopoietic stem cell transplantation has become the preferred treatment for patients under the age of 65 years. Prior to stem cell transplantation, these patients are given an initial course of induction chemotherapy. The most common induction regimens used today are thalidomide-dexamethasone, bortezomib-based regimens, and lenalidomide-dexamethasone [27]. Autologous stem cell transplantation-transplantation of a patient’s own stem cells after chemotherapy-is the most common type of stem cell transplantation for MM. It is not curative but does prolong overall survival. Allogeneic stem cell transplantation-transplantation of a healthy person’s stem cells into the affected patient-has the potential for cure but is available to only a small percentage of patients [28]. Furthermore, there is a 5%- 10% treatment-associated mortality rate. Patients over age 65 years and those with significant concurrent illness often cannot tolerate stem cell transplantation. For these patients, the standard of care has been chemotherapy with melphalan and prednisone. Recent studies among this population [29,30] suggest improved outcomes with new chemotherapy regimens. Treatment with bortezomib, melphalan, and prednisone produced an estimated overall survival of 83% at 30 months [31]; lenalidomide plus low-dose dexamethasone produced 82% survival at 2 years; and melphalan, prednisone, and lenalidomide produced 90% survival at 2 years. In other trials, lenalidomide plus high-dose dexamethasone proved to be superior to high-dose dexamethasone alone as treatment for newly diagnosed MM [32-34]. One study [35] looked at melphalan and prednisone plus thalidomide versus melphalan and prednisone versus VAD (vincristine, adriamycin, dexamethasone) induction, followed by high-dose melphalan and autologous stem cell transplantation in patients 65-75 years of age. The complete response rate was significantly better in the melphalan and prednisone plus thalidomide arm than in the melphalan and prednisone arm [35]. Melphalan and prednisone plus thalidomide are now recommended as first-line treatment. Melphalan and prednisone plus lenalidomide have also shown promise [35]. Other studies confirmed the superiority of adding thalidomide for prolonging survival in elderly, newly diagnosed patients with MM. Similar results were obtained with bortezomib [36,37].
Adjunctive therapy for MM includes radiation therapy to target areas of pain or an impending or existing pathological fracture. Bisphosphonates have a role in the secondary prevention of bony complications in MM, including hypercalcemia, pathological fracture, and spinal cord compression [38,39]. The American Society of Clinical Oncology (ASCO) issued a clinical practice guideline governing bisphosphonate therapy for MM patients who have lytic destruction of bone or a compression fracture of the spine from osteopenia [40]. ASCO recommends intravenous pamidronate, 90 mg delivered over at least 2 hours, or zoledronic acid, 4 mg delivered over at least 15 minutes every 3-4 weeks. Because the risk for osteonecrosis of the jaw is 9.5- fold greater with zoledronic acid than with pamidronate, patients may prefer pamidronate [40]. Zoledronic acid doses should be reduced in patients with preexisting mild to moderate renal impairment (estimated creatinine clearance 30-60 mL/min); the drug is not recommended for use in patients with severe renal impairment [41]. All patients receiving pamidronate or zoledronic acid therapy should be screened every 3-6 months for albuminuria. If unexplained albuminuria (>500 mg/24 hr) is found, ASCO recommends discontinuing the drug until the renal problems resolve [40]. Erythropoietin may ameliorate anemia resulting from either MM alone or from chemotherapy. It has been shown to improve quality of life [42,43]. In addition, one study demonstrated a survival advantage with the use of erythropoietin in patients with MM [44].
Renal failure in MM patients can be acute (reversible) or chronic (irreversible). ARF typically resolves when the calcium and paraprotein levels are brought under control. Treatment of Chronic Renal Failure (CRF) depends on the type of renal failure and may involve dialysis. Hydration (to maintain a urine output of >3 L/day), management of hypercalcemia, and avoidance of nephrotoxins (e.g., intravenous contrast medium, antibiotics) are also key factors. Smith AF, et al [45] suggested that bortezomib-based therapy can restore renal function in MM patients with renal failure. Bortezomib, however, has many adverse effects, including neuropathy, hypotension, and thrombocytopenia. Varicella zoster virus activation occurs in 10-60% of patients with MM treated with bortezomib. Antiviral prophylaxis (e.g., acyclovir 400 mg daily) has been found effective for preventing this activation [46-48]. In addition, the exact timing of bortezomib administration in the treatment plan of MM patients is still evolving through ongoing research. In an attempt to improve outcomes, direct removal of free light chains (FLCs) by plasma exchange has been studied [49-52]. A randomized controlled trial of 97 MM patients with ARF, however, failed to demonstrate any clinical benefit [53]. In that study, renal biopsies were not reported, serum FLC concentrations were not quantified, and most of the patients were not dialysis-dependent at presentation. Furthermore, plasma exchange does not result in sustained reductions in serum FLC concentrations, as demonstrated by clinical observations [54] and mathematical modeling. Leung N, et al. demonstrated that patients with cast nephropathy are more likely to recover renal function if a 50% decrease in serum FLC concentrations is achieved [55]. However, they failed to demonstrate any relation between the amount of plasma exchange and the degree of reduction in FLC concentrations or the renal response. To provide an alternative approach to plasma exchange for direct FLC removal, Hutchison CA, et al. [56] assessed the utility of extended Hemodialysis (HD), using a High-Cutoff (HCO) dialyzer. Detailed mathematical modeling showed HCO-HD to be far more effective than plasma exchange for FLC removal. In their pilot study, induction chemotherapy in combination with extended treatment by HCO-HD resulted in sustained reductions in serum FLC concentrations in most of their patients. These patients subsequently became independent of dialysis. The authors concluded that with dialysis-dependent ARF secondary to myeloma kidney, patients who undergo uninterrupted chemotherapy and extended HCO-HD are more likely to sustain reductions in serum FLC concentrations and to recover independent renal function. Resolution of cast nephropathy by HCOHD has been also supported by the report of Basnayake K, et al. [57].
Tumor Lysis Syndrome (TLS) is an oncological emergency characterized by a combination of metabolic disorders observed at the start of cancer treatment, or with the advancement of an aggressive malignancy. The syndrome is frequently associated with renal dysfunction, cardiac and skeletal manifestations, and gastrointestinal sequelae. TLS occurs with malignancies that are highly proliferative and have large tumor burdens, such as lymphomas and leukemias [58,59]. Metabolic abnormalities include hyperphosphatemia, hyperkalemia, hyperuricemia, and/or hypocalcemia. Renal dysfunction usually accompanies TLS. Often, hyperuricemia (generally, a uric acid level ≥ 476 μ mol/L) is a hallmark finding of TLS [58-64]. Adverse sequelae of TLS can precipitate life-threatening events, and if left untreated can lead to death. The prevalence of TLS varies among the hematological malignancies, and treatment-sensitive tumors, such as acute lymphocytic leukemia and Burkitt’s lymphoma, are associated with higher frequencies of TLS [58,59,61,65]. In studies on patients with intermediate- or high-grade NHL, abnormal laboratory results were more dramatic than the symptomatic clinical syndrome itself [66]. Silent laboratory evidence of TLS was seen in 70% of children undergoing induction chemotherapy, whereas significant symptomatic TLS occurred in only 3% [66]. As advances are made in the treatment of hematological malignancies, the incidence of TLS may increase [66]. Metabolic abnormalities and consequences Hyperkalemia may appear 6-72 hours after the initiation of chemotherapy [60-66] and is the most serious manifestation of TLS. Hyperkalemia must be corrected rapidly before potentially lethal ventricular arrhythmias occur. Hyperphosphatemia usually develops 24-48 hours following initiation of chemotherapy [59-67]. Calcium phosphate can precipitate when the solubility product of calcium and phosphate is exceeded, possibly leading to hypocalcemia. Muscle cramps, tetany, cardiac arrhythmias, and seizures can result [66,67]. In patients with myeloproliferative diseases or hematopoietic malignancies, nucleic acids are catabolized as a result of increased turnover of malignant cell populations. This results in an increase in purine metabolism, leading to hyperuricemia [58,59], which is usually apparent 48-72 hours following initiation of treatment [58,59]. Because of hyperuricemia, renal insufficiency develops when urine becomes supersaturated with uric acid, and crystals of uric acid form in the renal tubules and distal collecting system [68]. Despite management of metabolic abnormalities to reduce the risk of renal failure, 25% of children with advanced-stage Burkitt’s lymphoma and B-cell acute lymphoblastic leukemia still experience ARF secondary to severe hyperuricemia at the onset of chemotherapy [67,69]. In patients with TLS, the high uric acid load and hyperphosphatemia may overwhelm the ability of the nephron to autoregulate. The resultant decline in tubular flow may precipitate uric acid, leading to uric acid nephropathy. A urine uric acid/creatinine ratio of >1.0 is suggestive of uric acid nephropathy, whereas a ratio of <0.60-0.75 suggests renal failure of another etiology [58-62]. Patients with hematological malignancies are at increased risk of ARF from etiologies other than TLS. Acute tubular necrosis should always be considered in the differential diagnosis. Patients at risk of TLS are also susceptible to other forms of renal injury, owing to prolonged periods of hypotension or to exposure to nephrotoxic agents, such as antimicrobial agents, chemotherapy, or contrast media. In addition to the risk of nephrocalcinosis, damaged tubule cells may slough off into the lumens, resulting in obstructed nephron flow. These events further amplify the injury resulting from TLS-associated ARF [70,71]. Pathological studies demonstrate deposits of uric acid in the distal renal tubular lumens, causing intrarenal hydronephrosis. Uric acid crystals also can be seen in tubular epithelial cells and the medullary microcirculation [59,72,73].
The principles of management should address three critical areas: hydration, metabolic abnormalities, and supportive treatment of renal failure. Aggressive intravenous hydration not only helps correct electrolyte disturbances by diluting extracellular fluid, but it also increases intravascular volume. Increased volume enhances renal tubular flow, the glomerular filtration rate, and urine volume. Ideally, intravenous hydration with normal saline should begin 2 days prior to, and continue 2-3 days after, chemotherapy in high-risk patients [74,75].
Hyperkalemia: The major goals when treating acute hyperkalemia are cardiac membrane stabilization, intracellular shift of potassium, and reduction of the total potassium load. Acute treatment modalities include intravenous infusion of glucose plus insulin to promote redistribution of potassium from the extracellular space to the intracellular space, and intravenous calcium gluconate as cardioprotection for potassium levels >6.5 mmol/L or for those with electrocardiographic alterations. Intravenous hydration with alkaline fluid can also increase intracellular uptake of potassium. Potassium-wasting diuretics must be employed with caution, as they may worsen renal precipitation in the volume-contracted patient [76]. Long-term therapy such as oral potassium-exchange resins should be given immediately, because of the transient effectiveness of acute treatment modalities. If these measures fail to control serum potassium, hemodialysis should be initiated promptly. Dialysis prevents irreversible renal failure and other life-threatening complications. Indications for dialysis include persistent hyperkalemia or hyperphosphatemia despite treatment, volume overload, uremia, symptomatic hypocalcemia, and hyperuricemia [77,78]. Hemodialysis is preferred over peritoneal dialysis or Continuous Venovenous Hemofiltration (CVVHF), because of better phosphate, potassium, and uric acid clearance rates [77-79]. CVVHF has been used and is effective in correcting electrolyte abnormalities and fluid overload [78,79]. Because hyperkalemia can recur after dialysis is initiated and because of the high phosphate burden in some patients with TLS, electrolyte levels must be monitored frequently, and dialysis repeated as needed.
Hyperphosphatemia/hypocalcemia: Hyperphosphatemia is managed with oral phosphate binders and the same intravenous solutions of glucose plus insulin used for control of hyperkalemia. Hyperphosphatemia may lead to hypocalcemia, which usually resolves as phosphate levels are corrected. In some cases, depressed serum 1,25-dihydroxycholecalciferol levels contribute to hypocalcemia. Administration of calcitriol may correct the calcium levels. To avoid metastatic calcifications, however, such therapy should not be undertaken until serum phosphate levels have normalized [79].
Hyperuricemia: The standard treatment for hyperuricemia consists of allopurinol, urinary alkalinization, and hydration. Allopurinol blocks uric acid formation by inhibiting the enzyme xanthine oxidase [61,63,67]. Patients at high risk for TLS still need to excrete preexisting uric acid, which is not affected by allopurinol. Consequently, a drug that has rapid, highly potent uricolytic properties, reduces metabolic morbidity, and can be conveniently integrated with the initiation of a chemotherapy regimen would be helpful in this situation. Rasburicase is a recombinant form of urate oxidase enzyme available for this purpose [80]. A study in healthy adult male volunteers established that single daily intravenous doses of rasburicase were well tolerated and led to a dramatic decrease in plasma uric acid levels [81]. A multicenter Phase II trial of rasburicase in pediatric patients with leukemia and lymphoma (n=131) demonstrated that 100% of patients achieved uric acid control with rasburicase at a dose of 0.2 mg/kg, despite concomitant intensive chemotherapy [82]. Goldman SC, et al. presented a report demonstrating the efficacy of rasburicase compared with allopurinol in reducing uric acid levels in children with leukemia or lymphoma at high risk for developing TLS [83].
Kidney disease is ubiquitous in patients with hematologic malignancies, encompassing a wide spectrum of disorders involving each kidney compartment, including the vasculature, tubules, interstitium, and glomerulus, and there is significant overlap of kidney involvement with each hematologic malignancy. These renal disorders require prompt recognition by the clinician, due to the need to implement specific treatment to decrease the risk of subsequent chronic kidney disease.
None.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Al Dhneem HN, Al-Thwainy R, Al-Yahya MA, Abdul-Rahman IS (2023) The Association between Hematological Malignancies and Renal disease. Int J Nephrol Kidney Fail 9(2): dx.doi.org/10.16966/2380-5498.239
Copyright: © 2023 Al Dhneem HN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access