
Figure 1: The physiological structure of the brain and neurons in (a) healthy brain and (b) Alzheimer’s disease (AD) brain [2].
Thomas J Caputo1 Mohammed S Inayat2 Vincent S Gallicchio1*
1Department of Biological Sciences, College of Science, Clemson University, Clemson, SC USA, USA*Corresponding author: Vincent S Gallicchio, Department of Biological Sciences, 122 Long Hall, College of Science, Clemson University, Clemson, SC USA 29627, USA, E-mail: vsgall@clemson.edu
Alzheimer’s disease is characterized by progressive cognitive decline, including memory loss, impaired judgment, and changes in behavior. It is the most common cause of dementia and is known to generate pathological changes in the brain, such as amyloid plaques and neurofibrillary tangles which contribute to the symptoms. This disease affects over 55 million people worldwide and is known to frequently develop around the age of 65 and older. Bipolar disorder is characterized by extreme mood swings that include periods of mania involving heightened energy, euphoria, and depressive episodes leading to a loss of interest in many activities. These mood shifts can significantly impact the daily life and functioning of the affected person. There are more than 18 million people in the United States affected by this disease, and it is also known to be associated with the later development of neurodegenerative disorders like Alzheimer’s disease. Lithium has been used as a treatment for bipolar disorder for 60+ years while also being known to show effects decreasing the incidence of neurodegenerative disorders in a variety of patients. Regarding the treatment of Alzheimer’s disease, there is no present therapy approved to decrease the rate of development or cure the morbid disease after it has been diagnosed in individuals. Stem cell treatment is currently being studied through animal models to find a way to regenerate the loss of important neurological functions and combat the development of Alzheimer’s disease. Research has shown that lithium induces the proliferation of stem cells, including pluripotential stem cells and is a potent inhibitor of the biochemical process that results in the hyperphosphorlyation of Tau protein that is responsible for neurofibril tangles and neurological dysfunction observed in Alzheimer’s patients.
Alzheimer’s Disease; Stem Cell Treatment; Bipolar Disorder; Lithium
Aβ: β-amyloid; AD: Alzheimer’s Disease; ANK3: Ankyrin 3, APOE: Apolipoprotein E; APP: Amyloid Precursor Protein; BMMSCs: Bone Marrow Mesenchymal Stem Cells; BrdU: bromodeoxyuridine; CACNA1C: α-Calcium Channel Subunit; ChEIs: Cholinesterase Inhibitors; DG: Dentate Gyrus; EOAD: Early-Onset Alzheimer’s Disease; ESCs: Embryonic Stem Cells; GFP: Green Fluorescent Protein; GSK-3β: Glycogen Synthase Kinase-3 beta; Hup A: Huperzine A; IMP: Inositol MonoPhosphatase; IPSCs: Induced Pluripotent Stem Cells; LTCCs, L-Type Voltage-Gated Calcium Channel; MMSE: Mini-Mental State Examination; MSCs: Mesenchymal Stem Cells; NFTs: NeuroFibrillary Tangles; NMDAR: N-Methyl-D-Aspartate Receptor Antagonist; NSCs: Neural Stem Cells; PSEN1: Presenilin-1, PSEN2: presenilin-2; SGZ: Subgranular Zone; SP: Senile Plaques; SVZ: SubVentricular Zone
Alzheimer’s disease (AD) is a progressive neurological disorder that causes the degeneration of brain cells. The process of neurodegeneration is concurrent with the process of aging. Neurodegeneration or loss of structure or function of neurons can vary in severity based on the individual. The rate of progression, symptoms, age of onset, genetic factors, environmental factors, and overall health can affect its severity. Traumatic head injury, depression, cardiovascular and cerebrovascular disease, higher parental age, smoking, and family history of dementia are all known to increase the risk for individuals to develop this disease [1]. Alzheimer’s disease is the leading cause of dementia which is identified by a loss of intellectual thinking and declining memory resulting in less independence in personal daily activities [2]. The chances that an individual will develop Alzheimer’s disease doubles every 5 years after the age of 65. This chance based on age increases significantly from less than 1% per year before 65 years of age to 6% per year after 85 years of age. As individuals become even older, the rate increases from 10% after the age of 65 to 40 % after the age of 85 [1]. In 2019 Alzheimer’s disease was recorded to be the sixthleading cause of death in the United States and the seventh-leading cause of death in 2020 and 2021. The shift back in the rankings was only due to COVID-19 entering the top ten causes of death. However, AD has been an even higher cause of death among Americans aged 65 and older since 1995 [3-5]. The condition was finally recognized as a major public health problem in 1994 and was then added to the list of causes eligible to be ranked as leading causes of death in the United States [4]. Unpaid dementia care giving amounted to $339.5 billion in 2022, and increased family caregiver’s risk for emotional distress and negative mental and physical health [5]. More recently in 2023, long-term care and hospice services for people of ages 65 and older with dementia is projected to cost around $345 billion altogether [5]. Without intervention, these rates will continue to rise until an adequate treatment is found for AD.
On November 3rd, 1906, Alois Alzheimer, a German scientist, made an announcement that cemented his name into medical history [6]. The announcement included his discovery of abnormal symptoms that were generated in the cerebral cortex [6]. A patient he had been studying had symptoms including a progressive cognitive disorder, local neurological symptom, hallucination, delusion, and psychological social disability condition [6]. In 1984, the term probable Alzheimer’s disease was introduced to designate a clinically diagnosed acquired and progressive amnestic version of dementia that had no evidence for another etiology [7]. Probable Alzheimer’s disease characterized a clinicopathologic condition showing an intricate association between amnestic dementia and the presence of β-amyloid-containing neurotic plaques (Αβ plaques) and Tau-containing neurofibrillary tangles (NFTs) [7]. Alzheimer’s disease was previously regarded before the 1970s as a presenile dementia illustrated by Alzheimer’s original patient [7]. As of 2014 Alzheimer’s disease has been officially defined as a combination of amnestic dementia and biomarkers of amyloid pathology and “downstream topographical markers” consistent with neurotic plaque and NFT pathology [7].
The major theories currently related to physiological pathogenesis of Alzheimer’s disease involve the neuronal extracellular deposition of Aβ peptides which misfold into amyloid plaques and neuronal intracellular accumulation of hyperphosphorylated Tau protein to form NFTs (Figure 1), [8]. However, the major underlying factor for the cognitive and behavioral dysfunction observed in Alzheimer’s disease is synaptic dysfunction [9]. Both the cognitive and behavioral dysfunction seen in Alzheimer’s patients is mainly due to synaptic dysfunctions in the central nervous system [8]. Two primary types of neuropathological changes in Alzheimer’s disease that can explain the extent of disease progress and symptoms are positive and negative lesions [2]. Positive lesions can be characterized by an accumulation of neurofibrillary tangles, amyloid plaques, neuropil threads, and other deposits in the cerebral cortex of patients with Alzheimer’s disease [2]. Negative lesions can be characterized by neural, neuropil, a synaptic loss leading to substantial atrophy [2].
Figure 1: The physiological structure of the brain and neurons in (a) healthy brain and (b) Alzheimer’s disease (AD) brain [2].
Amyloid plaques are extracellular deposits containing Aβ protein that come in various morphological forms [2]. The transmembrane amyloid precursor protein (APP) is cleaved by proteolytic enzymes namely β-secretase and γ-secretase leading to the biosynthesis of Aβ deposits [2]. However, only a subset of the APP-immunoreactive neurites contained markers of the abnormal filaments characteristic of neurofibrillary pathology involved in Aβ plaques, otherwise known as senile plaques (SP) [9]. SP are 6- to 10-nm-wide filaments that contain 39- to 42-amino acid f3-amyloid proteins originating from the much larger transmembrane glycoprotein APP [9]. The specific size and morphology of these SP vary greatly depending on the region of their location [9]. Aβ42/43 is most found in Aβ plaques, and Aβ40, which tends to be more soluble, can be found later in the progression of the disease alongside blood vessels. The Aβ42/43 plaques found earlier in the disease can have APP fragments missing their C-terminus while the Aβ40 plaques found later can be found with Aβ42/43SPs [10].
NFTs are known to be one of the main pathological signs of AD which contain multiple types of hyperphosphorylated forms of the protein Tau [11]. The protein Tau was found to be essential for microtubule assembly [12]. The abnormal phosphorylation of Tau and resulting NFTs are thought to result from an imbalance of kinase and phosphatase activities [11]. In Tau, there is a significant amount of Ser/Thr-Pro motifs that can act as possible phosphorylation sites [13]. Because of the hyperphosphorylation possibility, the affinity of tau effectively binding to microtubules can be severely decreased [13]. In addition, because of hyperphosphorylation, Tau’s resistance to calcium‐activated neutral proteases can also be increased [14]. Paired helical filaments can aggregate because of the decline in affinity to the microtubules [15]. The influence of these processes on the Tau protein can finally lead to the formation into NFTs [14]. Specific sites on Tau as studied in AD brains including T231, S235, and S262 have some correlation with the clinical progression of AD [16]. NFTs resulting from the hyperphosphorylation of Tau proteins can clearly be noted to influence the formation of Alzheimer’s disease (Figure 2).

Figure 2: Formation of Neurofibrillary Tangles (NFTs) by the tau protein in tauopathies such as Alzheimer’s disease. Under pathological conditions, tau becomes hyper phosphorylated and detaches from microtubules. Phosphorylated tau then aggregates to form paired helical filaments (PHFs) and NFTs [17].
Genetics are known to play a role in the development of Alzheimer’s disease but are not solely responsible for its development in the way that other genetic disorders can be. Genetic research has concluded that the heritability of Alzheimer’s disease is about 70%, yet only 30% its heritability can be explained by genes known to be involved [17,18]. One common gene known to play a role in the development of Alzheimer’s disease is Apolipoprotein E (APOE). APOE, on chromosome 19, has three main alleles that affect the risk for Alzheimer’s disease being APOE ε4, ε3, and ε2 [19]. The APOE ε4 allele accounts for the highest increase of risk, the ε3 allele also accounts for increased risk, and the ε2 allele decreases the overall risk for Alzheimer’s disease [19]. In ε4 homozygotes, the frequency of Alzheimer’s disease is 91% and the mean age of at clinical onset is 68 which shows a strong correlation between this allele and the development of Alzheimer’s disease especially at an earlier age [19]. Amyloid protein precursor, presenilin-1 (PSEN1), and presenilin-2 (PSEN2) are other common genes that when mutated are known to cause autosomal dominant forms of early-onset Alzheimer’s disease (EOAD) (Figure 3) [20]. Mutated forms of these genes are found in 70% of families with more than one EOAD cases [19]. Recent developments using CRISPR-CAS9 have provided the possibility of applying Alzheimer’s disease genetic research results accurately and efficiently to new types of gene therapy [19]. Based on current genetic knowledge for Alzheimer’s disease, scientists are working to increase the amount of possible therapeutic targets.
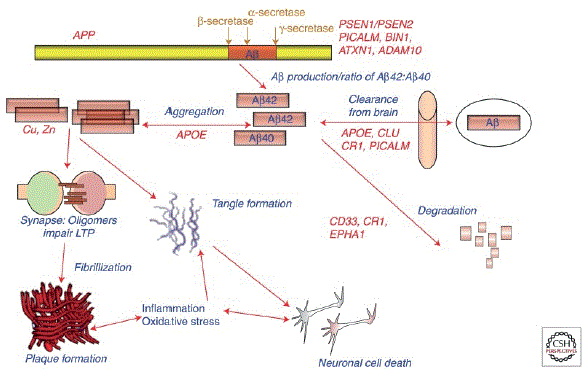
Figure 3: Potential roles of select Alzheimer disease (AD) genes in Aβ-related pathogenesis of AD [21].
Although Alzheimer’s disease has been recognizable and extremely prevalent for years, there are still little treatment options available. Currently, two classifications of pharmacologic therapy are recommended and available for Alzheimer’s disease patients: cholinesterase inhibitors (ChEIs) which include tacrine, donepezil, rivastigmine and galantamine and a N-Methyl-D-aspartate receptor antagonist (NMDAR) called memantine (Figure 4) [21-23]. Although effectively therapeutic to symptoms of Alzheimer’s disease, these medications do not prevent or cure Alzheimer’s disease [2]. Combination therapy has proven to be a flexible yet efficient way of providing therapy for this disease. The most common combinations include memantine and donepezil and memantine and galantamine which can act together to deliver synergistic neuroprotection for individuals with moderate to severe Alzheimer’s disease [24]. Another potential therapy, nitro-memantine, showed the ability to reverse the loss of brain connections and increase synapse prevalence to normal within only a few months of treatment in mouse Alzheimer’s diseasemodels [24]. Huperzine A (Hup A) is another type of ChE-I that was approved in China for treatment of Alzheimer’s disease in 1994 [25]. This ChE-I selectively inhibits acetylcholinesterase activity which increases acetylcholine levels in the brain to improve cognitive function [25]. This is an alternative treatment that has not yet been approved or regulated by the U.S. One non-pharmaceutical method associated with lower risk of various age-related diseases is adherence to a Mediterranean diet [26,27]. Ongoing studies on possible Alzheimer’s disease treatments show promise in attacking problems involving Aβ plaques and NFTs including hydro-methylthionine mesylate. This Tau aggregation inhibitor is shown to affect cognitive decline and brain atrophy in two Phase 3 trials in mild and moderate AD [28]. Another therapeutic antibody treatment, Donanemab, is currently being researched for reducing the number of amyloid plaques through binding to aggregated conformations of the Aβ42isomer [29]. The antibody slowed cognitive and functional decline in Alzheimer’s disease by 33% compared to placebo participants in Phase III trials, but also was said to have significant side effects present [29]. The major concern for future research is in which way to target the Αβ plaques and NFTs and at what time is clinical treatment relevant before disease progression. There is vast room for improvement in these treatments, and as a society, there needs to be a stronger interest taken in finding a cure to this disease. The magnitude of failed clinical trials makes it evident that scientists are either approaching this in an incorrect way or there needs to be some sort of innovation to come up with a better solution to this morbid disease.

Figure 4: Drugs approved for the treatment of Alzheimer’s disease: mechanisms of action, weight, and molecular structure.
An alternative treatment for Alzheimer’s disease currently being studied is stem cell treatment. Stem cells are undifferentiated cells that can mature into specialized cells and are classified by their origin and developmental potential. For instance, pluripotent stem cells can mature into specialized cell types from all three of the germ layers while multipotent stem cells can differentiate into a limited range of cell types within a particular tissue or organ (Figure 5) [30,31]. Induced pluripotent stem cells (iPSCs) are reprogrammed adult somatic cells that are now undifferentiated, pluripotent cells with the potential to become any cell type in the body and are like embryonic stem cells [31]. Mesenchymal stem cells (MSCs), which possess a multipotent nature and are derived from bone marrow, can transform into specific subtypes of glial cells responsible for producing myelin [31]. When these transformed cells are transplanted, they can offer essential support to injured neurons in the nervous system [31]. This approach creates a more favorable environment for natural neural recovery, thereby promoting regeneration within the nervous system [31]. Neural stem cells (NSCs) are another possible candidate for Alzheimer’s disease. When NSCs are transplanted, they have the potential to renew deteriorated cholinergic neurons, and new neurons originating from NSCs can establish synaptic links with nearby neurons [32]. The utilization of NSCs to replace and rehabilitate impaired cholinergic neurons and neural connections in the brain presents another theoretical avenue for exploring treatment possibilities for Alzheimer’s disease [32].

Figure 5: Types of stem cells commonly used to study and treat neurological deficits [31].
The possibilities of stem cell treatment for Alzheimer’s disease are currently being explored in animal clinical trials. Bone marrow mesenchymal stem cells (BMMSCs) have provided a possible therapy for patients with AD. BMMSCs were found to have improvement of memory loss and behavioral deficits in animal models with Alzheimer’s disease [33]. However, the mechanism for delivering the stem cell treatment is still full of complications and needs to be studied further. Mechanisms for delivery currently being studied include intravenous, intrahippocampal, intranasal, and intracerebroventricular delivery [34]. In another study, MSCs were found to enhance autolysosome formation in Aβ-treated cells giving a neuroprotective effect [35]. Overall, stem cell treatment seems like a viable option in the future for Alzheimer’s disease. The mechanism of delivery and side effects of treatment need to be further studied to find a safe yet effective way of delivering and treating Alzheimer’s disease.
In typical Alzheimer’s disease, brain atrophy initially affects the medial-temporal lobe and then spreads to the lateral-temporal and parietal cortices [36]. However, in atypical Alzheimer’s disease, atrophy tends to be most pronounced in areas related to the specific clinical symptoms, often not affecting the hippocampus in the early stages [36]. Impaired memory is one of the first and most prevalent signs of Alzheimer’s disease development though Alzheimer’s disease patients may develop severe cognitive and behavioral symptoms like depression, anxiety, anger, irritability, insomnia, and paranoia as the disease continues to progress [1]. Different Alzheimer’s disease phenotypes can include various symptoms such as visual or motor difficulties in 8-13% of cases, language difficulties in 7-9%, and executive dysfunction in 2% while sex-based variations in the presentation of symptoms note that women are more commonly affected by visual/spatial and motor symptoms and behavioral symptoms more prevalent in men (Figure 6) [36]. In addition, some individuals may experience overlapping symptoms.

Figure 6: MRI across AD phenotypes; blue arrows indicate atrophy [36].
Bipolar affective disorder is a mental health condition characterized by extreme mood swings between manic and depressive episodes. This illness is linked to high rates of premature mortality from both suicide and medical comorbidities and is a leading cause of disability [37]. Bipolar disorder can be classified under two distinct subtypes: I and II. A bipolar I disorder diagnosis requires the presence of mania, and bipolar II disorder requires hypomania with at least one major depressive episode (Figure 7) [38]. Bipolar II tends to be more common than bipolar I although clinical trials have more frequently attempted to treat bipolar I disorder [38]. Studies have suggested there is a lifetime prevalence of about 1% for bipolar type I in the general population, while another survey of 11 countries concluded that the lifetime prevalence of bipolar spectrum disorders was 2.4%, with a prevalence of 0.6% for bipolar type I and 0.4% for bipolar type II [37]. This chronic illness is typically experienced first in early adulthood with smaller, yet possible chances of onset in childhood or in older age [38]. Depression is comorbid with bipolar disorder and may impose a greater weight of both duration and impact when seen with bipolar disorder compared to manic symptoms [38]. The most common age of onset for bipolar disorder is around the early twenties [37]. There has been a bimodal distribution of the frequency of bipolar disorder suggested, where there are two peaks in age of onset around 15-24 years old and around 45-54 years old [37]. Bipolar disorder in the United States had an annual cost estimated at $45 billion with $7 billion from direct costs for inpatient and out-patient care and $38 billion from lost productivity of wage earners, homemakers, individuals who were in institutions or who had committed suicide, and caregivers [39]. Most patients undergo treatment for this disorder that normally includes pharmacologic therapy, including lithium medications [38]. Misuse of substances (cannabis, opioids, cocaine, sedatives, and alcohol) and causality has been suggested in both directions regarding bipolar disorder comorbidity [37]. Although the significant comorbidity is unquestionable, establishing causality becomes more challenging due to the elusive nature of determining the timing connection between substance misuse and the onset of mental illness [37].

Figure 7: Polarity of symptoms for bipolar disorder subtypes [40].
The term “bipolar” dates to 1957 where a psychiatrist, Karl Leonhard, first coined the term to classify patients with both manic and depressive symptoms [40,41]. In 1980, the Diagnostic and Statistical Manual of Mental Disorders used Leonhard’s term “bipolar” to replace their term “manic-depressive disorder” [42]. Pharmacological tools in clinical practice were not very efficient in the field of mental pathology until the discovery of lithium salt’s antimanic effect in the 1950-60s [43]. In the late 1960s in Europe, lithium was first found effective in treating manic episodes from bipolar disorder. In the United States the FDA first authorized lithium for anti-maniac treatment in 1970 and for the treatment of manic-depressive episodes in 1978 [43]. New antipsychotic drugs have been introduced and authorized by the FDA since the early 2000s like olanzapine, risperidone, quetiapine, and ziprasidone for anti-maniac treatment and drugs including quetiapine and lurasidone for antidepressant treatment [43]. In addition, an antiepileptic agent called lamotrigine was authorized for prescription in 2003 to prevent depressive episodes in bipolar disorder [43]. Before treatment using these authorized drugs, chemical substances like chloral hydrate and bromides were used to treat psychotic and manic patients in the 19th and early 20th centuries [43]. Treatment has ranged over the past century; however, lithium salt treatment has proven effective in treating bipolar disorder for over sixty years now and remains the most effective prescribed medical treatment for bipolar disorder [43].
It is extremely common for the initial diagnosis of bipolar disorder to be missed as it often hides its true nature at the beginning, and it may take several years before the condition is correctly identified [44]. The typical progression of this mental condition is characterized by recurrent manic and hypomanic events, which can include periods of depressive states [44]. Because of the elusiveness of this disease, scientists are currently researching possible indicators that point towards bipolar disorder, so patients can be diagnosed and treated earlier. First and foremost, one of the main known contributions to bipolar disease is genetic with twin studies suggesting monozygotic concordance of between 40-70% [37]. Genetic factors will be further discussed in the next section. With the use of functional MRIs and other neuroimaging techniques, a possible marker is an overreaction malfunction in ventrolateral prefrontal cortex along with an increased level of hippocampus and amygdala emotion-processing [45]. This leads to typical bipolar symptoms including extreme changes in emotion and increases overall sensitivity [45]. When looking at this indication structurally, it was found that gray matter volume decreases in the prefrontal and temporal cortices, the amygdala, and the hippocampus along with a fractional anisotropy decrease in white matter tracts within the connection between prefrontal and subcortical regions [45]. Studies in patients with bipolar disorder have consistently shown abnormalities in peripheral inflammatory markers, particularly elevated levels of soluble interleukin-2 receptor, soluble interleukin-6 receptor, tumor necrosis factor-alpha, and interleukin-4 [46]. However, researchers are still uncertain whether these abnormalities are mood state-dependent or inherent to the disease process [46]. Further research that considers co-occurring medical factors contributing to inflammation is needed to better understand the role of inflammation in bipolar disease. Additionally, multiple neurotransmission systems have been implicated in the pathophysiology of bipolar disorder. The serotonin and norepinephrine systems, as well as the dopamine system, have shown associations with mood disorders like bipolar disorder which is supported by neuroimaging studies and genetic associations [47]. The GABA and glutamate systems have been considered, as well, with proposed connections from studies including neurochemical, genetic, and neuroimaging studies [47]. Although scientists are aware of these possible factors, the exact reasons for the development of this disorder are still unclear and are being researched to this day.
Inheritance of bipolar disorder is a multifaceted process that involves a genetic predisposition, complex polygenic inheritance, and interactions with various environmental factors [48]. The two main genetic markers that are suspected in being associated with the development of bipolar disorder are ankyrin 3 (ANK3) and the α-calcium channel subunit (CACNA1C) [49]. One study tested over 1.8 million variants of the specific locus with over 4,000 cases and 6,000 controls and found these two regions in strong association with the bipolar disorder cases [50]. CACNA1C encodes a subunit of the L-type voltage-gated calcium channel (LTCCs), and with a possible altered regulation using intron 3, methylation of CpG islands can affect this Ca+ channel possibly leading to development of bipolar disorder [51]. The mechanism of action of lithium is like that of calcium channel blockers which leads some scientists to believe lithium treatment could be useful in fixing the altered regulation of LTCCs due to the CACNA1C gene [52]. Genetic factors alone do not determine the development of bipolar disorder, and scientists must attempt to continue to understand other factors to be able to determine a more comprehensive view of the disorder’s etiology
Bipolar disorder is normally seen in patients for the remainder of their life once diagnosed; therefore, it requires lifelong treatment strategies. One of the major medications used to treat bipolar disorder is lithium. Lithium is known to be exceptional in treating both Type I and Type II bipolar disorder along with solely treating mania [53]. Furthermore, lithium has a unique potential to display anti-suicide effects in patients which is not only beneficial because of the significant relevance of high suicide rates in patients with bipolar disorder, but also specific unto lithium compared to other drugs used to treat this disorder [53]. In recent history, antipsychotics like Olanzapine and anticonvulsants have been more frequently prescribed to treat bipolar disorder, but they are known to be associated with metabolism issues and a slight sedation effect [54]. There have been reports indicating that lithium demonstrates not just neuroprotective properties preserving brain structures, but also contributes to volumetric expansion associated with emotional regulation [55]. For instance, it affects areas like the prefrontal cortex, hippocampus, amygdala, anterior cingulate cortex, subgenual anterior cingulate cortex, inferior frontal gyrus, and postcentral gyrus [55]. Similarly, lithium is also known to correspond with the formation of stem cells in the subventricular zone, striatum, and forebrain, which could explain this said volumetric increase [55]. The initial application of lithium to treat a bipolar patient was in a 1954 study by Mogens Schou [56]. Schou gave a bipolar patient in his care lithium to keep his mood swings from being too drastic [56]. Later in 1970, Schou presented his findings that lithium had a clear preventive effect in mood disorders [56]. Currently, many atypical antipsychotic drugs are used in monotherapy to treat bipolar disorder, but other cases lithium treatment is frequently seen in combination with anticonvulsant medications to treat this disorder along with depression [57]. One study detailed that their results indicated a risk of any relapse to be 36% for lithium and 61% for Placebo over the course of one year which shows an impressive absolute risk reduction of 25% [58]. Some studies suggest that there are some possible negative side-effects including renal toxicity and cognitive problems which should be considered before prescribing lithium. However, in recent years, these negative effects have been refuted showing that if lithium treatment is applied in a safe, clinical manner, it can be one of the best treatment options for bipolar disorder [54,59,60].
Alzheimer’s disease and bipolar disorder are both complex neurological medical conditions that are influenced by numerous unknown genetic variants [61]. Alzheimer’s disease is a neurodegenerative disorder while bipolar disorder is a neurodevelopmental disorder [61]. There are mass amounts of significant clinical information that suggests a relationship between this disorder and disease. Research on patients with bipolar disorder has revealed a heightened risk of receiving a dementia diagnosis compared to other individuals in the general population of similar gender and ages. Additionally, for each hospitalization due to a bipolar disorder episode, the risk of a dementia diagnosis increases by 6% [62]. A casecontrol study in Western Australia that was based on comprehensive hospital records found that individuals aged 65-84 with bipolar disorder had double the likelihood of developing Alzheimer’s disease compared to patients without the disorder [63]. Furthermore, using MRI technology, scientists have been able to analyze changes in neural tissue properties associated with Alzheimer’s disease over the aging process [64]. These specific changes could prove useful to researchers in helping to understand Alzheimer’s disease and in providing a possible location for interventional therapeutic treatment [64]. The hippocampus is known to play a key role in memory formation and storage in the human brain. In Alzheimer’s disease, neurodegeneration significantly reduces the hippocampal volume as seen in brain MRIs [65]. Neuroimaging of the hippocampus in bipolar brains suggests a reduction in hippocampal volumes, as well [66]. The same study also notes that patients who had been taking lithium to treat bipolar disorder did not have that same degradation of the hippocampus in neuroimaging tests [66]. Research is currently underway to establish a clearer connection between hippocampal volume reduction and bipolar disorder, as these two conditions often appear to be associated especially in older individuals [65,67]. Overall, there seems to be a link between bipolar disorder and Alzheimer’s disease in old age which I will examine further when considering possible treatments for these medical conditions.
Developing stem cell technology has enhanced the properties of self-renewal, growth, specialization, and adaptability of stem cells making it possible to transform them into various types of specialized neurons and glial cells within the central nervous system [68]. These advancements have led to successful results in animal models of Alzheimer’s disease [68]. There are four possibilities for the major types of stem cells that could possibly be used in Alzheimer’s disease treatment being NSCs, MSCs, embryonic stem cells (ESCs), and iPSCs. This review discusses the two most studied and effective types in possible Alzheimer’s disease treatment: MSCs and NCSs [69].MSCs are classified as pluripotent cells and can be obtained from umbilical cord blood, the Wharton jelly, bone marrow, and adipose tissue [68]. They inhibit the activation of inflammatory microglia while encouraging the activation of anti-inflammatory microglia helping prevent ongoing tissue damage from chronic neuroinflammation [70]. Additionally, MSCs facilitate the accumulation of microglia around Aβ deposits to enhance clearance, and they may also promote autophagy activation, which could be responsible for clearing Aβ-plaques [70] (Figure 8). In one study, intracerebral injections of various types of MSCs were given to samples of mice [71]. Following transplantation, synaptic activity increased, and spatial learning and memory were enhanced [71]. Aβ-plaque deposits were diminished and Tau hyperphosphorylation was reduced as the MSC treatment was able to increase various Aβ degrading enzymes and decrease several proinflammatory cytokines [71]. Overall, MSC treatment diminishes neuroinflammation by eliminating Aβ-plaques, disentangling NFTs, and addressing irregular protein degradation [69]. MSC therapy fosters the restoration of the blood-brain barrier and autophagy processes, maintains acetylcholine levels, and enhances cognitive function in the brain [69]. If researched further, MSCs could be a potential candidate for human clinical trials to eventually become a treatment for AD.

Figure 8: Mechanism of protective effects of mscs in AD [70].
NSCs are self-renewing, multipotent stem cells that can transform into various cell types, including neurons and glial cells [72]. NSCs can be obtained from brain tissue or genetically reprogrammed from somatic cells [72]. They can be primarily found in the subgranular zone (SGZ) of the dentate gyrus (DG) and the subventricular zone (SVZ) of the lateral wall of the ventricle [73]. Transplanting NSCs that secrete growth factors has enhanced neurogenesis and cognitive function in rodent Alzheimer’s disease models [34]. In rodent models affected by alkaline neurotoxicity, the transplantation of human NSCs with high choline acetyltransferase expression has been seen to reverse deficits in spatial memory and learning [68]. In other studies, NSCs are shown to have the potential to protect cerebral capillaries, reduce Aβ and tau pathologies, decrease inflammation, and enhance neurogenesis (Figure 9) [32]. Another study further demonstrated that transplanted human NSCs in the brains of mice effectively reduce Tau phosphorylation levels [74]. NSCs are another type of stem cell that has great potential to develop into a treatment for neurodegenerative disorders like AD if studied more extensively.

Figure 9: Neural stem cells therapy for Alzheimer’s disease [73].
Lithium remains a primary choice for treating bipolar disorder, addressing acute episodes and long-term management, due to its established effectiveness in managing mania, reducing suicidal tendencies, and preventing relapses [75]. While the exact mechanisms of its mood-stabilizing effects are not fully understood, emerging research suggests that lithium may also support neuroprotection and neuroplasticity [75]. Monovalent lithium can compete with bivalent magnesium for substrate binding sites due to their similar ionic radii which disrupts enzymes relying on magnesium as a cofactor [76]. Consequently, lithium can inhibit various important enzymes, including glycogen synthase kinase-3 beta (GSK-3β), inositol monophosphatase (IMP), and Akt/β-arrestin [76]. GSK-3β is found predominantly in the brain which makes lithium treatment a potential candidate for modifying disease progression in conditions like Alzheimer’s disease, as it inhibits enzymes related to neurological pathways relevant to neuropsychiatric and neurodegenerative disorders [76]. GSK-3β has been identified as a molecular link to hyperphosphorylation of Tau (Figure 10). When inhibited in mice trials, results show that there is a slower neurodegeneration process as both neuronal cell death decreases, and tau is phosphorylated at the normal, decreased rate [77]. If lithium provides a possible mechanism for inhibiting GSK-3β, it could prove to be invaluable in producing a treatment for neurodegenerative disorders including AD.

Figure 10: Overexpression of GSK-3β induces tau-dependent AD-like pathology [80].
Neurodegenerative disorders such as dementia and more specifically Alzehimer’s disease are currently affecting over 55 million people worldwide [78]. There is presently no cure for this medical condition, although several of the most promising possibilities for potential developing treatments have been discussed. Nevertheless, when used over a long period of time and often seen with treating bipolar disorder as mentioned above, lithium is seen to have antineurodegeneration capabilities. In a 2006 clinical study in Japan, Terao et. al., compared Mini-Mental State Examination (MMSE) scores of 30 patients receiving lithium and 25 controls with 35 patients with lithium prescription history and 20 controls [79]. They found that patients previously prescribed lithium had significantly better MMSE scores compared to controls compared to the patients just now receiving the lithium treatment compared to the controls [79]. This suggests a potential positive impact of lithium on dementia prevention when it has been used chronically over a lifetime. In a 2007 clinical study in Brazil, Nunes et. al., focused on bipolar disorder patients and found that the prevalence of AD was significantly lower in the group receiving continuous lithium treatment (5%) compared to the group without lithium treatment but with other mood-stabilizers (33%) [80]. Multinomial logistic regression analysis confirmed an association between lithium use and a reduced risk of AD, suggesting that lithium may have a protective effect [80]. They noted that this protective effect could be due to the impact of potentially direct inhibition of GSK-3 by lithium [80]. In a 2010 epidemiological study in Denmark, Kessing et al., analyzed data from 4,856 patients with bipolar disorder wherein 2,449 had been previously exposed to lithium treatment [81]. During the follow-up period which was after the 10-year study from 1995 to 2005, only 216 patients were diagnosed with dementia [81]. The data yet again shows a possible correlation in lithium treatment and the prevention of dementia and Alzheimer’s disease [81]. A retrospective cohort study was conducted using clinical records from Cambridgeshire and Peterborough NHS Foundation Trust, UK, from 2005 to 2019 [82]. The study included 29,618 patients aged 50 or older without prior mild cognitive impairment or dementia. Out of them, 548 were exposed to lithium [82]. The exposed group had lower dementia risk, including Alzheimer’s and vascular dementia, compared to the unexposed group, even after adjusting for various factors [82]. Lithium appeared to provide protective effects for both short-term and long-term users, with some evidence of increased benefit for longer use [82]. After analyzing multiple studies like the ones above, it is concluded that there is most likely a connection between chronic lithium uses and the prevention of neurodegenerative disorder whether it be through inhibition of GSK-3 or another related pathway.
Because of the known association between lithium and stem cells, the combined effects of stem cell treatment along with lithium therapy was explored. Lithium, recognized for its GSK-3β inhibitory properties and possible neuroprotective and regenerative effects, could potentially influence the activities of MSCs. One study used bromodeoxyuridine (BrdU) incorporation assays along with green fluorescent protein (GFP) tagged MSCs to observe the effect of different lithium doses on stem cell proliferation [83]. They recorded that lithium at a concentration of 0.1 mM enhanced cell viability and the number of BrdU-positive cells in GFP-MSCs [83]. This data suggested that lithium promotes MSC proliferation at specific lowdose concentrations [83].
Another study discovered that lithium could enhance the in vitro proliferation of human BMMSCs [84]. At a concentration of 5 mM, Lithium treatment increased cell growth without causing cell death which they linked to an increase in the proportion of cells in the S phase and the expression of cyclin D1 [84]. In addition, they noted that the mechanism involved the inhibition of GSK-3β by lithium [84]. Overall, the study suggested that lithium can boost MSC proliferation via inhibiting GSK-3β which could potentially enhance the effectiveness of MSC transplantation therapy [84]. In a 2018 study, Zhang et al., analyzed the effect of a lithium chloride therapy on the proliferation of NSCs [85]. They found that the lithium treatment suppressed GSK3β levels leading to extreme stimulation of the proliferation of NSCs [85]. From these studies, one can see a similar positive effect of lithium treatment on the proliferation of both MSCs and NSCs. However, there needs to be more research done on this topic to be able come to a definite conclusion supporting the combination of stem cell therapy and lithium therapy. When researching potential neurodegenerative disorder treatments, this combination shows a great deal of prospective promise for possible future treatments.
Lastly, it was plausible to consider when the proposed possible treatment could be applied clinically and when it could possibly be promoted as an option to proactively decrease the chances of neurodegenerative disease for prospective patients. EOAD is much rarer as it is seen in less than 10% of Alzheimer’s disease patients, thus the focus looked toward the more typical age range of the start of development from around 60 to 70 years [1]. If chronic lithium therapy can eventually be used as therapy as suggested by previously mentioned studies [79-82], a small yet effective dosage could be considered up to 10 years prior to the typical age range of onset in prospective individuals to possibly protect against neurodegeneration. This would allow the lithium treatment to be already in the patient’s system acclimated and ready in advance to aid with the prevention of neurodegeneration. Regarding prospective individuals, this would include groups that have extensive genetic histories of Alzheimer’s disease and dementia to be examined before the clear effect of genetics on the development of Alzheimer’s disease. Stem cell therapy would only be applicable when neurodegeneration begins as they are a limited resource and will not fight the neurodegeneration process if it has not begun. When signs of neurodegeneration begin, stem cell treatment, possibly using MSCs or NSCs, could be applied in a method that would have to be researched as effective in previous animal trials. This treatment would ideally reverse the neurodegeneration process and continue to differentiate aiding the formation of replacement neurons for those previously lost. Also, if prospectively combined with previous chronic lithium treatment, these stem cells would have the potential for increased proliferation to aid the reversal of the neurodegeneration process faster and more effectively as seen in above studies [83-85]. For trials testing these possible future treatments, it would be recommended to have multicentered, double-blinded, randomized tests using placebos and eventually applying a range in varying doses of both lithium and stem cells in respective treatment options. Overall, in the future if these prospective treatments are clinically studied and approved, a proactive approach to attacking neurodegenerative disorders would be extremely beneficial in combating this ever-present medical condition and could lead to a better quality of life worldwide.
The frequency of Alzheimer’s disease is becoming increasingly more relevant regarding its overall worldwide effect. AS this neurodegenerative disorder becomes more and more prevalent, there is still no real, effective means of curing or effectively treating it today. AD patients can most likely attribute the development of their disease to the increased amount of Aβ plaques and hyperphosphorylation of tau leading to neurofibrillary tangles along with possible hereditary factors. A combination of these ailments leads to neuronal degeneration including a loss of synaptic function and cell death. If further developed, injections of stem cells such as MSCs and NSCs could prove to be extremely beneficial in guiding neurodegeneration and reversing the loss of neurons along with increasing the hippocampal volume to where it should be in a healthy individual. When looking at the incidence between Alzheimer’s disease and bipolar disorder, one can see a clear connection between the two medical conditions. This allows us to consider treatments like lithium therapy as an alternative to treating both conditions. Chronic lithium use in patients exemplifies a clear connection with a decreasing chance of neurodegeneration as patients become older by inhibiting the GSK-3β protein. In addition, the prospective combination of both lithium and stem cell treatment, if researched further, could yield an effective treatment for both proactive and active measures in treatment for reversing the neurodegenerative capabilities of AD. In conclusion, stem cell treatment and lithium, especially with chronic use, presents a possible effective approach to therapy for Alzheimer’s disease. However, there is no question that these therapeutic measures, although they show great future potential, need additional research and clinical trials before proving definitively effective and being released to the public world as a form of therapy for hindering Alzheimer’s disease.
The authors have no conflict of interest to declare.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Caputo TJ, Inayat MS, Gallicchio VS (2024) Potential Stem Cell Treatment for Alzheimer’s Disease with Possible Applications of Lithium Therapy. J Clin Lab Med 7(1): dx.doi.org/10.16966/2572-9578.142
Copyright: © 2024 Caputo TJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access