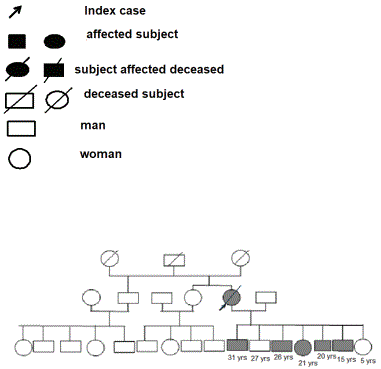
Figure 1: Genetic Family Tree 1.
Guillaume Mahamat Abderraman1* Ali Mahamat Hissein1 Brahim Soukaya2 Djidita Hagre Youssouf3 Toglengar Adneli Alliance1 Yousra Aboul Bashar1 Charfadine Senoussi3 Tara Fotclossou1
1Department of Nephrology-Dialysis, Renaissance University Hospital Center, N’Djamena, Chad*Corresponding author: Guillaume Mahamat Abderraman, Department of Nephrology-Dialysis, Renaissance University Hospital Center, N’Djamena, Chad, Tel: 00235.66.61.95.95; E-mail: zalba2001@yahoo.fr
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic renal disease. After about ten years of evolution, it leads to a slow and progressive loss of kidney function eventually causing renal insufficiency and kidney failure. Few data exist on this pathology in Africa and Chad. The objective of this study was to analyze the epidemiological, diagnostic and therapeutic aspects of ADPKD.
Methodology: This was a multicenter cross-sectional, descriptive and analytical study conducted over a period of 34 months in two hospitals in N’Djamena, Chad. All patients aged 15 years and above, having a family history of polycystic kidney disease, which were hospitalized or who were coming for consultation that met the unified criteria for ultrasonographic diagnoses (Ravine criteria modified by Pei) of ADPKD were included in the study. The clinical, paraclinical, therapeutic and evolutionary criteria for these patients had been studied.
Results: There were a total of 26 cases of polycystic kidney disease that had a hospital prevalence of 1.16%. The average age was 42.4 years with extremes ranging from 15-70 years and a sex ratio of 1.3. The family survey had shown that parental consanguinity was present in 34.6% (n=9). The prevalence of hypertension was 53.8% (n=14). The mean serum creatinine was 45 mg/l with extremes ranging from 4.5 to 274.42 mg/l. It was noted that 65.3% (n=17) had chronic kidney disease with a GFR >30 mL/min/1.73m2. During diagnosis 26.9% (n=7) had end-stage renal disease (ESRD), while 2 patients progressed to ESRD two years after diagnosis. One patient was hospitalized for chronic hemodialysis. No patient had received a kidney transplant and no patient had undergone a molecular biology and genetics study or had been treated with Tolvaptan.
Conclusion: In Chad, the hospital prevalence of ADPKD was 1.16%, affecting young adults (average age of 42.4 years) with a male predominance. Management is usually late and remains limited to symptomatic treatment with complications such as chronic renal failure.
Abbreviations: ADPKD: Autosomal Dominant Polycystic Kidney Disease, CRF: Chronic Renal Failure, ESRD: End-Stage Renal Disease, CVA: Cerebro Vascular Accident
Autosomal dominant polycystic kidney disease; Chronic kidney disease; Chad
Autosomal dominant polycystic kidney disease is a genetic disease which leads to a slowly progressive evolution towards chronic kidney failure that causes cysts to form on the kidneys that are scattered throughout the renal cortex and medulla. This can start at any time in life and is sometimes associated with extra renal manifestations. It is one of the most common genetic diseases affecting 100,000 people in France and 12 million worldwide [1]. The clinical severity of polycystic kidney disease is extremely variable [2]. After about ten years, the natural evolution is towards kidney failure. End-stage chronic renal failure is observed in 50% of cases from the age of 50 and above. In Africa, studies on ADPKD are few. In Senegal, Yaya K reported a prevalence of 0.75% [3]. In Chad, no clinical study on autosomal dominant polycystic kidney disease had been carried out. This is why we initiated this work whose objectives were to analyze the epidemiological, diagnostic and therapeutic aspects of ADPKD.
This was a multicenter, cross-sectional, descriptive and analytical study conducted over a period of 34 months from 1st January, 2018 to 1st October, 2020. The study took place in N’djamena, in the Nephrology Department of the Renaissance University Hospital and the National Reference University Hospital. These two are tertiary hospitals with a hospital capacity of 240 beds and 410 beds, respectively. All patients aged 15 years and above, having a family history of polycystic kidney diseases, which were hospitalized or who were coming for consultation that met the unified criteria for ultrasonographic diagnoses of ADPKD were included in the study (Table 1). The diagnosis of autosomal polycystic kidney disease was retained on the basis of the presence of a family history of polycystic disease and on ultrasound arguments (Ravine criteria modified by Pei, family history, or genetic testing).
All patients presenting on ultrasound with polycystic kidneys that did not meet the unified criteria for ultrasonographic diagnoses of ADPKD were excluded from the study. The clinical, paraclinical, socio-demographic and therapeutic data had been collected. This study had made it possible to draw up the genealogical tree of certain families. Data analysis and processing were performed using Microsoft Excel and a SPSS software version 18.0. The descriptive results will be expressed as a percentage and the analytical results will be significant if the p value is less than 0.05. All patients had given an informed consent and authorizations from the hospital administration and ethics committee were obtained.
During our study, a total of 2229 patients had consulted from both hospitals. Fifteen cases met the unified criteria for ultrasonographic diagnoses of ADPKD, which makes a hospital prevalence of 0.67%. The family history of polycystic kidney disease in these patients had led to the establishment of their different family as mentioned in the figures 1-3 (3 families). This research identified 11 patients were diagnosed as carriers of the polycystic kidney disease gene. The average age was 42.4 years with extremes ranging from 15 to 70 years with a sex ratio of 1.3. The age group between 40 to 60 years predominated with 46.2% (n=12). The discovery of polycystic kidney disease was fortuitous in 11.5% (n=3). The family survey had shown that parental consanguinity was present in 34.6% (n=9). The average age of onset of the first clinical signs was 37.7 years. Hypertension was present in 53.8% (n=14), followed by abdominal pain, urinary tract infection and chronic renal failure, 7.6% (n=2); 7.6% (n=2) and 3.8% (n=1), respectively. Grade 1, 2 and 3 hypertensions were found in 42.1% (n=8); 36.9% (n=7) and 21% (n=4), respectively. On physical examination, back pain in the lumbar region was present in 61.5% (n=16) and hepatalgia in 3.8% (n=1). It was noted that 15.3% (n=4) had macroscopic hematuria requiring an average hospitalization of 2 days with a blood transfusion of whole blood. One of the infectious complications present were urinary infections which accounted for 19.2% (n=5). The cytobacteriological examination of urine had shown a bacterial infection of Escherichia Coli in 12% (n=3) of cases. Other complications were summarized in figure 4.
Figure 1: Genetic Family Tree 1.

Figure 2: Genetic Family Tree 2.

Figure 3: Genetic Family Tree 3.

Figure 4: Distribution of patients according to complications.
Biologically, the average hemoglobin level was 10.87 g/dl with extremes ranging from 4.5 to 14 g/dl. A hemoglobin level between 8 to 10 g/dl was found in 57.7% (n=15) of cases. The mean serum creatinine level was 45 mg/l with extremes ranging from 4.5 to 274.42 mg/l. On admission, 65.3% (n=17) had chronic kidney disease with a GFR >30 ml/min. Patients that had reached end-stage renal disease was present in 26.9% (n=7). None of the patients had undergone a molecular biology and genetics study. On abdominal ultrasound, 61.5% (n=16) of patients had enlarged kidneys and 84.6% (n=22) had more than 6 cysts per kidney. The distribution of patients according to ultrasound findings is shown in figure 5.
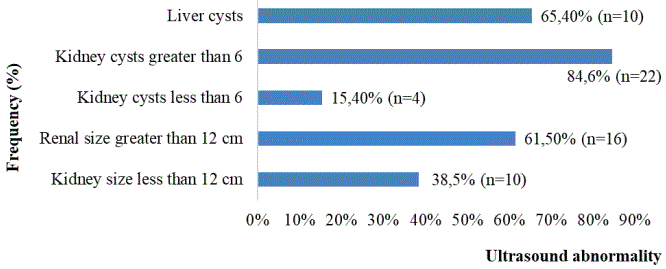
Figure 5: Distribution of patients according to ultrasound data.
Evolution was marked by the slow progression towards kidney failure, which was present in 7.7% (n=2). This had increased the total of patients with end-stage renal disease from 6 to 9 patients (34%). There was a significant relationship between the incidence of chronic kidney disease, severity of hypertension, severity of the urinary tract infection and the presence of stroke (p=0.034). 78% (n=19) of patients were hypertensive and were put on antihypertensive drugs such as angiotensin-converting enzyme (ACE) inhibitors. None of the patients were treated with desmopressin or tolvaptan. One patient was admitted for chronic hemodialysis. No patient had undergone a kidney transplant.
Renal polycystosis is a rare genetic pathology with a major prevalence around the world. In Europe, the prevalence of ADPKD was estimated to be 3.96 people out of 10,000 while in France 4.59 out of 10,000 [4]. Ackoundou-N’Guessan C, et al. [5] and Yaya K, et al. in Senegal [3] found a prevalence of 1/500 and 1/333, respectively, compared to 1/131 in Chad. The high rate of consanguineous marriage and polygamy could explain this high rate compared to the other countries, which was demonstrated by a pedigree. This family tree history seeks the notion of positive heredity based on the certainty of the existence of renal cysts or the notion of family dialysis, stroke or death from renal failure in the family. In our study, the family nature of the disease could be established in 50% of the patients, a result close to that found in Nadjoua, which was 57% [6]. The average age of patients was 42.4 years compared to 51 years found by Nadjoua in 2011 in Morocco [6]. High blood pressure secondary to polycystic kidney disease affects men and women in the same proportions. Hypertension is noted in almost all patients in our study. It is a major modifiable cardiovascular risk factors but also a factor in the progression of chronic kidney disease. The diagnosis of hypertension is made early and at a younger age compared to patients with essential hypertension [7,8]. In our study, the familial nature of the disease was established in 50% of patients, a result close to that found in Najoua with 57% of cases [6]. The sex ratio was different from other studies which showed a female predominance [3,5]. In polycystic kidney disease, hypertension is a very frequent and early symptom according to Harrap SB, et al. [9]. In our series, hypertension was noted in 73% of patients. It was present in all patients in Kane’s series [9]. The cardiovascular morbidity and mortality of patients with ADPKD is significant. This is both linked to chronic renal failure and to the particular cardiovascular phenotype of these patients, which is characterized by an increased prevalence, compared to the general population with hypertension [10]. Hematuria is one of the suggestive signs of this condition [11]. It was found in 15.3% (n=4) of cases. Kidney size was determined by abdominal ultrasound in all patients. The association of hepatic and renal cysts seems to increase with age and the degree of renal insufficiency. In our study, 64% (n=17) had asymptomatic hepatomegaly. This result is confirmed in the literature where more than three quarters of patients with polycystic kidney disease developed hepatic cysts. Among them, only 10 to 20% develop symptomatic hepatomegaly [12]. In our study, back pain in the lumbar region was present in 34.6% compared to 80% in Madagascar [13]. One patient had a stroke. In polycystic kidney disease, several studies have shown an acceleration of the pulse wave velocity, indicating an increase in vascular stiffness which appears very early in the history of the disease [14]. All patients in the study were put on ACE inhibitors. After 3 years of follow-up, these patients had a stable renal function. A randomized and controlled study conducted by Torres VE, et al. on “Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease” had demonstrated that sodium restriction is beneficial in the management of ADPKD. This study suggests that a salt-restricted diet associated with the use of angiotensin converting enzyme inhibitors may have a slightly favorable effect on renal volume, improvement in glomerular filtration rate and on the mass index of the left ventricle [15]. In our study, the severity of CKD was correlated with grade 3 hypertension and the occurrence of stroke. Vascular stiffness measured by pulse wave velocity is a predictive marker of chronic kidney disease progression in patients with ADPKD [16]. More than half of the patients had a GFR >30 ml/min/1.73m2 and more than 60% (n=15) had an increased renal volume. The criteria for starting Tolvaptan (a vasopressin V2-receptor antagonist) is a GFR >30 ml/min/1.73m2; significant nephromegaly associated with the risk of a loss of renal function; signs of rapid disease progression such as the presence of clinical manifestations (renal pain, hemorrhage, intracystic infection, macroscopic hematuria) or a significant loss of GFR of at least 5 ml/min by year as described by Buxeraud J [17]. This molecule was approved by the regulatory authorities in Japan (March 2014), Health Canada (February 2015), and the European Medicines Agency Committee for Medicinal Products for Human Use (May 2015) as a therapy for patients with ADPKD and CKD stages 1 to 3 with evidence of a rapidly progressing disease [18,19]. Tolvaptan slowed the rates of total kidney volume (TKV) growth and renal function decline over a 3 year period in patients with ADPKD that were enrolled in the Tolvaptan Efficacy and Safety Management. Total kidney volume and GFR are markers of ADPKD progression, with TKV used [20,21].
In Chad, hospital prevalence of ADPKD was 1.16% affecting young adults with an average age of 42.4 years and a male predominance. Management is usually late and remains limited to symptomatic treatment of complications such as chronic renal failure.
To all the co-authors who actively participated in this original research work.
The authors declare no conflicts of interest.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Mahamat Abderrraman G, Mahamat Hissein A, Soukaya B, Hagre Youssouf D, Adneli Alliance T, et al. (2022) Autosomal Dominant Polycystic Kidney Disease: An Unknown Disease in Chad. Int J Nephrol Kidney Fail 9(1): dx.doi.org/10.16966/2380-5498.234
Copyright: © 2022 Mahamat Abderrraman G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access