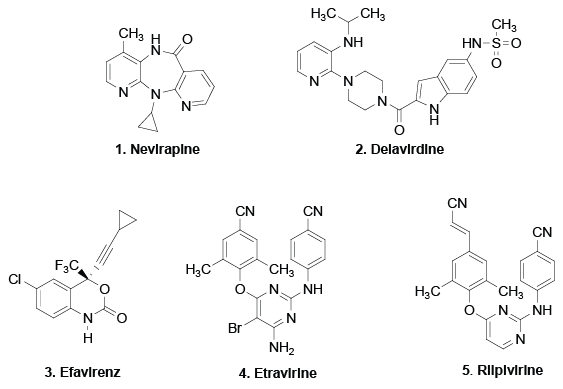
Figure 1: Structures of currently approved NNRTIs.

Yang Ge Jiasheng Xu Zhendong Song Xiaodong Ma*
College of Pharmacy, Dalian Medical University, Dalian, P.R. China*Corresponding author: Xiaodong Ma, College of Pharmacy, Dalian Medical University, Dalian, 116044, P.R. China, Tel:+86-0411-86110419; Fax:+86-0411-86110419; E-mail: xiaodong.ma@139.com
Developing non-nucleoside HIV-1 reverse transcriptase inhibitors (NNRTIs) with improved drug resistance profiles is still a great challenge owing to the rapid emergence of resistant virus. For the potential activity against both wild-type and clinically relevant NNRTI-resistant mutant HIV-1 strains, the novel NNRTI benzophenone derivatives (BPs) have drawn great attention. Considerable efforts on modifying this template have led to the identification of the most active BP analogue, GW678248, which has been progressed to the phase II clinical studies owing to its high anti-HIV-1 activity, low clearance and stable metabolic property. Therefore, this review focused on the benzophenone deravitives provided valuable SAR information for further modifying this scaffold.
HIV; NNRTI; SAR; Benzophenone; Inhibitor
Since the identification of the human immunodeficiency virus (HIV) as the causative agent of AIDS, searching for more safe and effective drugs for HIV therapy continues to be a hot topic [1,2]. Till now, 27 approved anti-HIV drugs have been marketed for the treatment of HIV infection, which have dramatically alleviated the AIDS issues particularly after the application of highly active antiretroviral therapy (HAART) [3-6]. Being the most important components of HAART, the marketed non-nucleoside inhibitors of reverse transcriptase (NNRTIs), nevirapine (1, NVP) [7], delavirdine (2, DLV) [8], efavirenz (3, EFE) [9-11], etravirine (4, ETV) [12,13] and rilpivirine (5, RPV) [14-17], which exhibited great resistance to the presence of drug resistance mutations, have been proved effective for the AIDS therapy (Figure 1). Nevertheless, due to the rapid emergence of drug resistance, more effective HIV inhibitors are still required to allow continued suppression of the viral infection [18-20].
Recently, benzophenone derivatives, originated in a high-throughput screening in 1995 [21], attracted more attention for their greatly improved profiles of activity against both wild-type and clinically relevant NNRTIresistant mutant strains of HIV-1. Structural modifications on this backbone have resulted in producing several novel BP analogues, such as 7 (GW4511) [22] and 8 (GW678248) [23,24], which possess high antiHIV activity, low clearance and stable metabolic property (Figure 2). In particular, GW678248 also has been advanced to the clinical exploration for the excellent potency in inhibiting the mutant HIV strains (IC50<1 nM) [25]. As our great interest, we have designed and synthesized a series of new potential BP inhibitors, such as PAFAs, NPEs, and so on [26-29]. Based on these studies, the structure and activity relationships (SARs) of BPs were summarized and provided for further structure optimizations in this manuscript.
Apparently, the shape of A-ring has a significant impact on the potency of the BP analogues. As shown in table 1, phenyl-substituted analogue 9a not only showed nanomolar concentration level of anti-HIV activity assayed in MT-4 cell, but also exhibited equivalent potency against syncytia (SYN) and p24 antigen (p24) in C8166 cells infected with HIV- 1. Replacement of the A-ring with a more polar pyridyl ring (9b) or the saturated cyclophenyl ring (9c) resulted in a great loss of anti-HIV activity [21].
By moving a small substituent around the A-ring, compounds 10a-d were obtained and evaluated for the activity against both the wild-type and the mutant HIV viruses (Table 2) [23]. Clearly, placement of a small group at meta position (10c) is far more favorable than the substitution of the same group at ortho (10b) or para position (10c). Furthermore, substitution at the meta position (10c) offers an advantage over the unsubstituted analogues (10a), particularly against the critical mutant viruses K103N and Y181C.
Figure 1: Structures of currently approved NNRTIs.
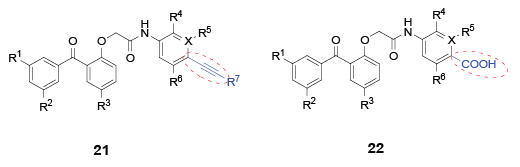
Figure 2: Novel benzophenone NNRTI structures.
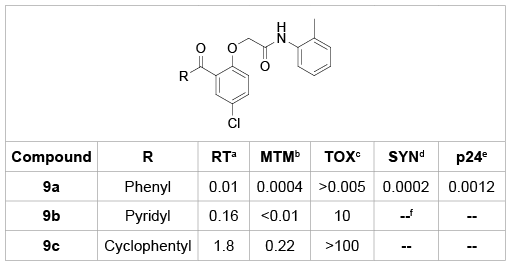
Table 1: BP analogues with various A-ring and their anti-HIV activity.
a
IC50 versus RT of HIV-1 (µg/mL), AZT triphosphate control=0.02 ± 0.005
µM;
b
IC50 versus HIV-1 (strain RF)-infected MT-4 cells (µg/mL), AZT=0.002–
0.02 µM;
c
IC50 versus uninfected MT-4 cells (mg/mL); d IC50 versus syncytia formation in infected (C8166-cells µg/mL),
AZT=0.002–0.02 µM;
e
IC50 versus p24 antigen formation in infected (C8166-cells µg/mL),
AZT=0.001–0.01 µM;
f Undetectable.
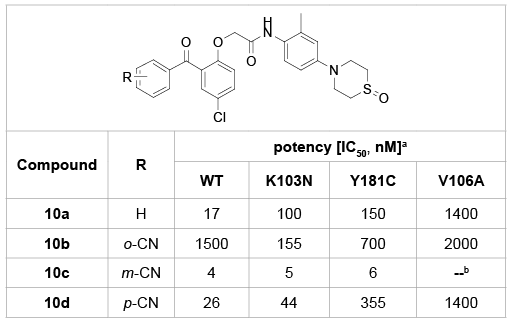
Table 2: SARs of various substituents on the A-ring.
a
IC50 values determined using a HeLa-MAGI assay. Wild-type virus (WT)
was the HxB2 strain. K103N, Y181C, and V106A mutants were isogenic
with the HxB2 virus backbone;
b Undetectable.
Romines et al. [23] further explored the meta substitution patterns on the A-ring (Table 3). Addition of a methyl group (11h) or a chloro group (11i) at the second meta position on the A-ring of the mono-meta substituted analogue (10c), had remarkable impact on the ability of the analogue to inhibit the V106A strain, without sacrificing the potency in the wild-type, K103N, and Y181C viruses. The disubstituted analogues shown in table 3 have very good profiles against the wild-type, K103N, Y181C, and V106A viruses, including the fluoro, trifluoromethyl (11b), the chloro, bromo (11f), and the chloro, methyl (11g). Although all of these were more potent against the V106A strain than the mono-meta substituted analogue, they were not quite as potent as the methyl, cyano analogue (11h). Use of electron-donating substituents (11c) was clearly less beneficial than the electron withdrawing substituents, although potency against the Y181C mutant virus was less affected than potency against the other viruses in the panel. Use of neutral substituents (methyl groups), however, was very well tolerated. In fact, the dimethylsubstituted analogue (11d) was as potent as the methyl, cyano-substituted analogue (11h).
Three versions of the thiophene B-ring analogues 12a-c were prepared and evaluated for the activity against wild-type HIV in MT4 cells (Table 4) [23]. Compared with the chlorophenyl B-ring (9a, IC50=10 nM), the thiophenyl compound 12a showed approximately 30-fold decrease of activity. Although the activities of compounds 12b and 12c were improved, their potency was low relative to the analogues with the chlorophenyl B-ring (9a).
In 2011, Ma et al. [27] obtained a series of benzophenone derivatives (13a-h) with B-ring substituted by a naphthyl ring to increase the π–π interactions with the mutant type of HIV viruses (Table 5). The results showed that most of these compounds were highly effective, and the most active analogue 13d was able to inhibit HIV-1 replication within 4.9 nM concentrations. However, these compounds are still less sensitive to the A17 mutant HIV viruses (micromolar level). Although the naphthyl B-substituted analogues were not as potent as the lead compound, they provided a new scaffold worthy of further optimizations.
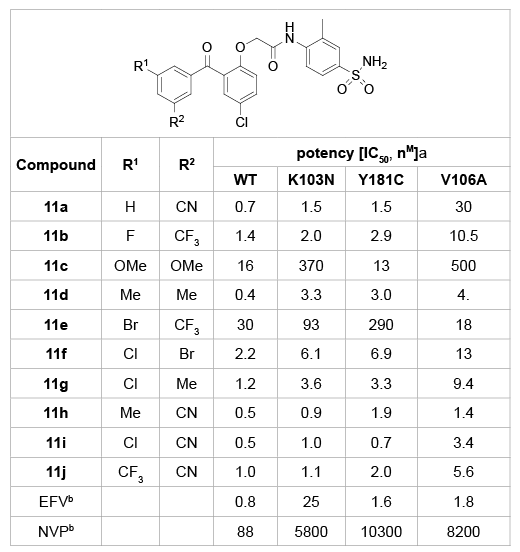
Table 3: SARs of 3,5-disubstituents on the A-ring.
a
IC50 values determined using a HeLa MAGI assay. Wild-type virus (WT)
was the HxB2 strain. K103N, Y181 C and V106A mutants wereisogenic with
the HxB2 virus backbone;
b
EFV is efavirenz; NVP is nevirapine.
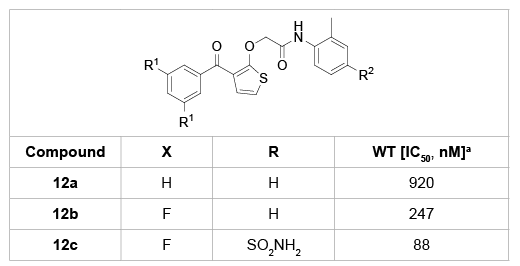
Table 4: BP analogues bearing a thiophenyl B-ring and their anti-HIV
activity
a
IC50 values were determined using HIV-1 IIIB wild-type virus in an acute
infection assay in MT4 cells.
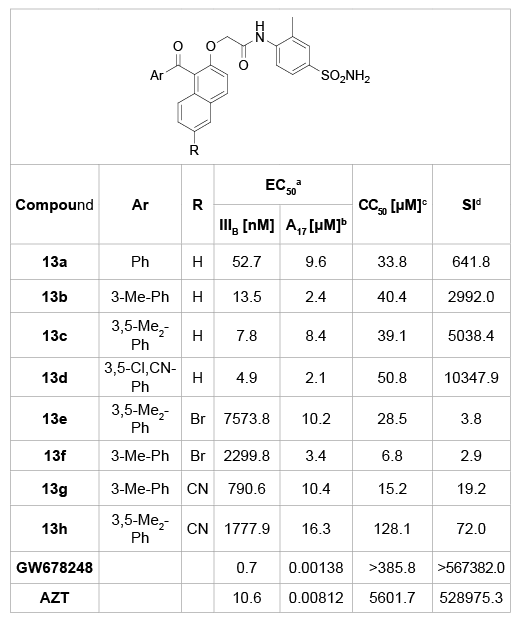
Table 5:BP analogues bearing a naphthyl B-ring and their anti-HIV activity.
a
EC50: effective concentration of compound required to protect the cell
against viral cytopathogenicity by 50%in C8166 cells;
b
A17: HIV-1 mutated strain bearing both K103N andY181C mutations;
c
CC50: cytotoxic concentration of compound that reduces the normal
uninfected C8166 cell viability by 50%;
d
SI: selectivity index: ratio CC50/EC50 (HIV-1 IIIB).
As illustrated in table 6, compounds 14a-f were designed and synthesized to investigate the SARs of substitutions on the B ring [21]. Obviously, 5-chloro substituted 14a was as potent as 5-fluorine substituted 14b in inhibiting HIV viruses assayed in both RT and cell level. Replacement of chlorine with a polar imidazole group (14c) resulted in a significant loss of activity. The dichloro-substituted compounds 14d-f also showed significant SAR information that the 4,5-dichloro-substituted 14e retained the activity of the mono substituted whereas the 3,3- and 5,6-dichloro derivatives 14d,f were devoid of activity.
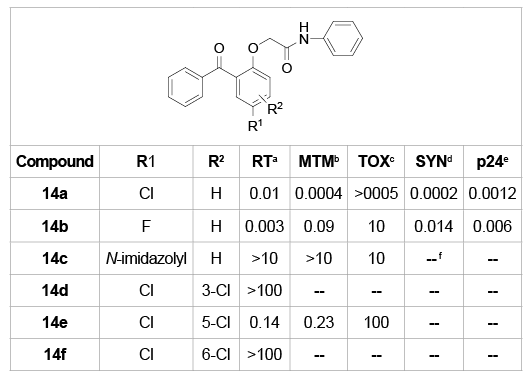
Table 6: SARs of various substituents on the B-ring
a
IC50 versus RT of HIV-1 (µg/mL), AZT triphosphate control=0.02 ± 0.005
µM;
b
IC50 versus HIV-1 (strain RF)-infected MT-4 cells (µg/mL), AZT=0.002–
0.02 µM;
c
IC50 versus uninfected MT-4 cells (mg/mL); d IC50 versus syncytia formation in infected (C8166-cells µg/mL),
AZT=0.002–0.02 µM;
e
IC50 versus p24 antigen formation in infected (C8166-cells µg/mL),
AZT=0.001–0.01 µM;
f
Undetectable.
Ma et al. [28] further explored the importance of the carbonyl group between A and B-rings in 2011 (Table 7). By reducing the carbonyl group, they synthesized a series of benzhydrol derivatives 15a-h and evaluated their anti-HIV activity in C8166 cells. Among these molecules, most were able to inhibit wild-type HIV-1 at lower than 1 µM. In particular, compound 15d was identified as the highest active inhibitor against the wild-type HIV-1, with an EC50 value of 0.12 µM and a selectivity index value of 312.73. Although some of them exhibited moderate activity against the double mutant strain A17 (K103N+Y181C), these inhibitors were still not quite sensitive to the resistant profiles. Taken together, this exploration induced a valuable conclusion that the benzophenone carbonyl is essential for BPs to maintain their high anti-HIV potency.
Also based on GW678248, Tucker et al. [30] changed the keto group between A and B-rings into an ether linker, to synthesize a series of diaryl ether derivatives 16a-d which possess broad antiviral activity against a number of key clinical mutations. As shown in table 8, adding the 3-chloro substituent to the central aryl ring (16a) increased the activity versus WT and the Y181C mutant RT enzyme. Addition of a meta cyano substituent to the A-ring resulted in a large enhancement of potency (16c) versus WT and both the K103N and Y181C enzymes, and the further addition of a second meta chloro substituent provided a more moderate enhancement in potency versus the K103N mutant (16d). This di meta substitution on the A-ring appears to provide excellent potency versus WT and the key mutant enzymes, which is similarly to the SAR seen with GW678248.
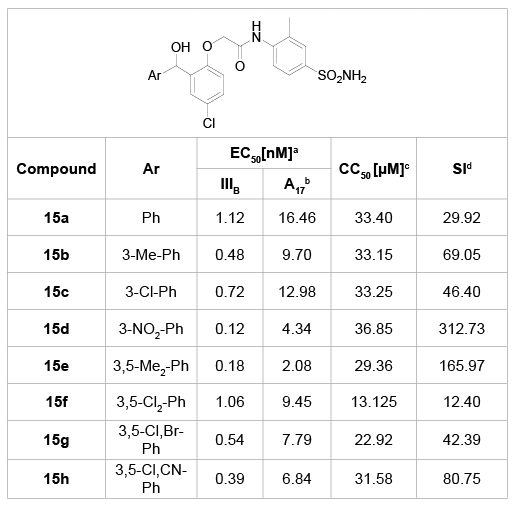
Table 7: Benzhydrol derivatives 15a-h and their anti-HIV activity
a
EC50: effective concentration of compound required to protect the cell
against viral cytopathogenicity by 50% in C8166 cells;
b
A17: HIV-1 mutated strain bearing both K103N andY181C mutations;
c
CC50: cytotoxic concentration of compound that reduces the normal
uninfected C8166 cell viability by 50%;
d
SI: selectivity index: ratio CC50/EC50 (HIV-1 IIIB).
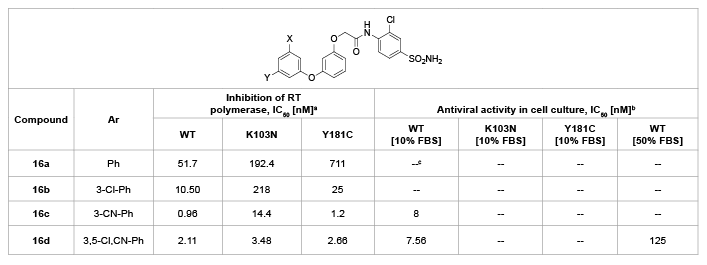
Table 8: Diaryl ether derivatives 16a-d and their anti-HIV activity
a
Compounds were evaluated in a standard SPA assay. Values are the geometric mean of at least two determinations;
b
CIC95 (Cell culture inhibitory concentration) is defined as the concentration at which the spread of virus is inhibited by >95% in MT-4 human T-lymphoid
cells maintained in RPMI 1640 medium containing either 10% FBS or 50% NHS. Values are the geometric mean of at least two determinations. No
cytotoxicity was observed for any of the compounds up to the upper limit of the assay (8.3 µM);
c
Undetectable.
Inspired by the active diaryl ether HIV inhibitors, Gu et al. [29] designed a series of naphthyl phenyl ether analogues (NPEs, 17a-i, Table 9), in which the bulky naphthalene ring was introduced to replace the benzene ring (A ring). The evaluation for their activities against HIV-1 in C8166 cells showed that most of the compounds moderately repress the replication of HIV-1 virus. In particular, compound 17f showed the highest activity against the wild-type HIV-1 with an EC50 value of 4.60 nM, along with the moderate activity against the double mutant strain HIV-1 A17 (K103N+Y181C) and HIV-2 strain ROD, with EC50 values of 0.82 and 4.40 µM, respectively. Unfortunately, this structural modification led to about 5-fold decrease of anti-HIV activity compared with GW678248.
In 2011, Ma et al. [26] synthesized a class of N-phenylaryl formamide derivatives (PAFAs, 18a-i, Table 10), considering that the flexible amido linker between A and B rings might not only improve the adaptation of the inhibitor to RT, but also promote the H-bond bindings with key mutant amino acids Y188 or Y181. Within these inhibitors, more than half possess anti-wild-type HIV-1 EC50 values ranging from 0.3 nM to 5.1 nM, and the selectivity index values ranging from 10616 to 271000. In particular, compound 18c (EC50=0.30 nM, SI=184578), 18f (EC50=0.37 nM, SI=212819), 18g (EC50=0.32 nM, SI=260617) and 18h (EC50=0.27 nM, SI=271000) displayed the highest activity against this type virus as potent as GW678248. Moreover, the four compounds also were effective to inhibit the double mutant strain A17 (K103N+Y181C) with EC50 values of 0.29 µM, 0.14 µM, 0.10 µM and 0.27 µM, respectively. Interestingly, compound 18g, the broad-spectrum anti-HIV inhibitor, also showed potent activity against HIV-2 ROD with an EC50 value of 4.37 µM.
Wyatt et al. [21] first explored the preliminary SARs of the C-ring in 1995. As shown in table 11, the N-cyclohexylamide 19a lost the total activity against several mutant HIV viruses compared with the phenylsubstituted 19b. Although the 2-methylbenzyl amide derivative 19c was less active than 19b in enzyme and cellular assays, it was significantly more metabolically stable. The bicyclic amide 19f synthesized as a constrained version of 19c proved to be a potent inhibitor of the enzyme and exhibited excellent activity in antiviral assays, but was less metabolically stable than 19c. Also, compound 19e with (N,N-diethylamino)-propoxy substituent on the C-ring have the significant metabolic stability problem. All these results indicated that the anilino aromatic ring was essential for their potential anti-HIV activity, while the 2-sustituted methyl of C-ring increased the metabolic stability of BPs.
By replacing the phenyl ring (C-ring) with a pyridine ring, Romines et al. [23] synthesized a family of pyridine-substituted BP derivatives (20a-g) and evaluated their anti-HIV activity (Table 12). Apparently, the sulfonamide substitution at the pyridine ring was quite well tolerated (20c-g, IC50<10 nM). Placement of the pyridine nitrogen at the X3 position (20g) attenuated potency against both wild-type (IC50=18 nM) and K103N mutant viruses (IC50=93 nM), while installing the pyridine nitrogen at either the X1 (20c,d) or X2 (20e,f) positions gave analogues with excellent potency against both wild-type and mutant viruses with IC50 values ranging from 1 to 10 nM.
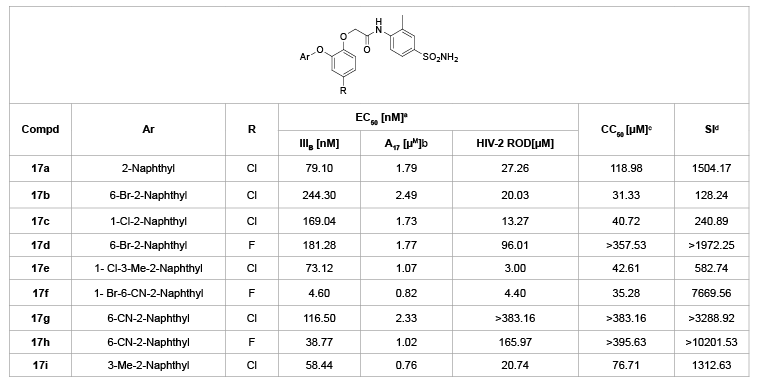
Table 9: Naphthyl phenyl ether analogues 17a-I and their anti-HIV activity
a
EC50: effective concentration of compound required to protect the cell against viral cytopathogenicity by 50%in C8166 cells;
b
A17: HIV-1 mutated strain bearing both K103N andY181C mutations;
c
CC50: cytotoxic concentration of compound that reduces the normal uninfected C8166 cell viability by 50%;
d
SI: selectivity index: ratio CC50/EC50 (HIV-1 IIIB)
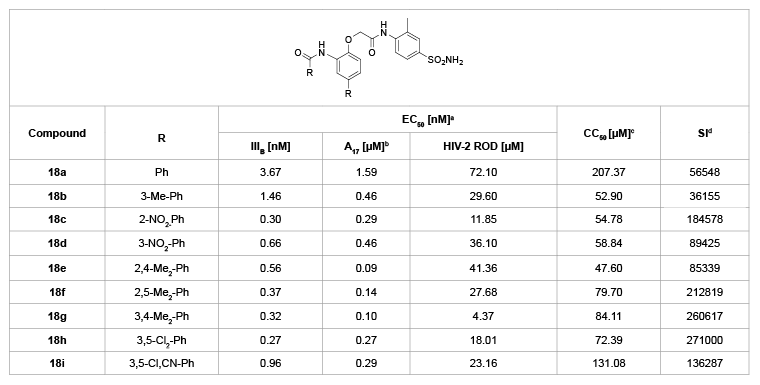
Table 10: N-Phenylaryl formamide derivatives 18a-i and their anti-HIV activity
a
EC50: effective concentration of compound required to protect the cell against viral cytopathogenicity by 50% in C8166 cells;
b
A17: HIV-1 mutated strain bearing both K103N and Y181C mutations;
c
CC50: cytotoxic concentration of compound that reduces the normal uninfected C8166 cell viability by 50%;
d
SI: selectivity index: ratio CC50/EC50 (HIV-1 IIIB)
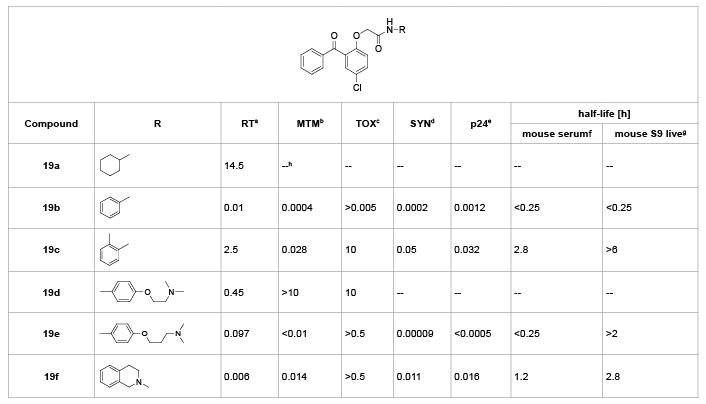
Table 11: SARs of C-ring.
a
IC50 versus RT of HIV-1 (µg/mL), AZT triphosphate control=0.02 ± 0.005 µM;
b
IC50 versus HIV-1 (strain RF)-infected MT-4 cells (µg/mL), AZT=0.002–0.02 µM;
c
IC50 versus uninfected MT-4 cells (mg/mL);
d
IC50 versus syncytia formation in infected (C8166-cells µg/mL), AZT=0.002–0.02 µM;
e
IC50 versus p24 antigen formation in infected C8166-cells µg/mL L), AZT=0.001–0.01 µM;
f
Stability of compound in mouse serum preparation;
g
Stability of compounds in mouse S9 liver preparation;
h
Undetectable.
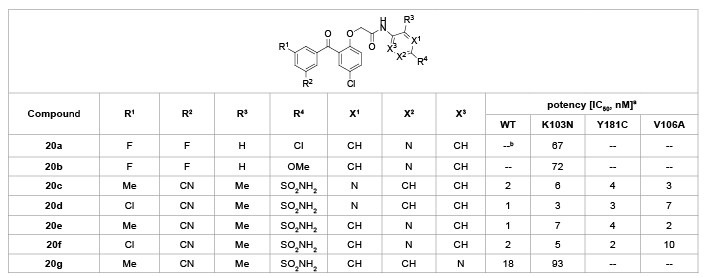
Table 12: SARs of various substituents on the C-ring.
a
IC50 values were determined using a HeLa MAGI assay. Wild-type virus (WT) was the HxB2 strain. K103N, Y181C, and V106A mutants were isogenic
with the HxB2 virus backbone;
b
Undetectable.
In 2006, a family of potent HIV inhibitors 21-22 featuring a para-substituted alkynyl or carboxyl group on the C-ring were reported [31,32]. All these compounds were able to inhibit the wild-type and the single or the double mutant HIV strains within nanomolar concentrations. Aquino et al. [33] further explored the SARs of the para substituent on the C-ring, and obtained three chiral benzophenone analogues 23-25. All the three chiral molecules exhibited less than 10 nM potency against the wild-type and a panel of mutant HIV strains. Excitingly, their concentrations for 50% of maximal effect on the key mutant strains, such as K103, Y181 and Y188, were even lower than 1 nM (Figure 3).
As shown in table 13, various para-substituents on the C-ring were employed to further investigate the SARs of BPs [23]. Conversion of the ether linkage to secondary amines or amides (26b-e) was little benefit to these changes relative to the unsubstituted analogue 26a. Yet, compound 26e is quite potent against both wild-type (IC50=5 nM) and K103N mutant viruses (IC50=9 nM). Changing the terminal amine with other groups proved to be more consistently beneficial. The sulfonamide (26f), urea (26g), and ether (26h) substituents were all favorable in the antiviral assays. The best results were achieved when the para position was substituted with a sulfonamide (26i). This analogue had excellent potency against both wild-type virus and the two key mutant viruses associated with NNRTI resistance (K103N and Y181C) with an EC50 value of lower than 3 nM concentrations. Analogues with the sulfonamide substitution (26j,k) were also active in the antiviral assays, but were not quite as potent as 26i. In general, substitution at the para position was beneficial. These explorations also indicated that most of the compounds did not show notable difference in potency (p<0.05), even in cases with fairly significant changes (e.g., 26b vs 26i).
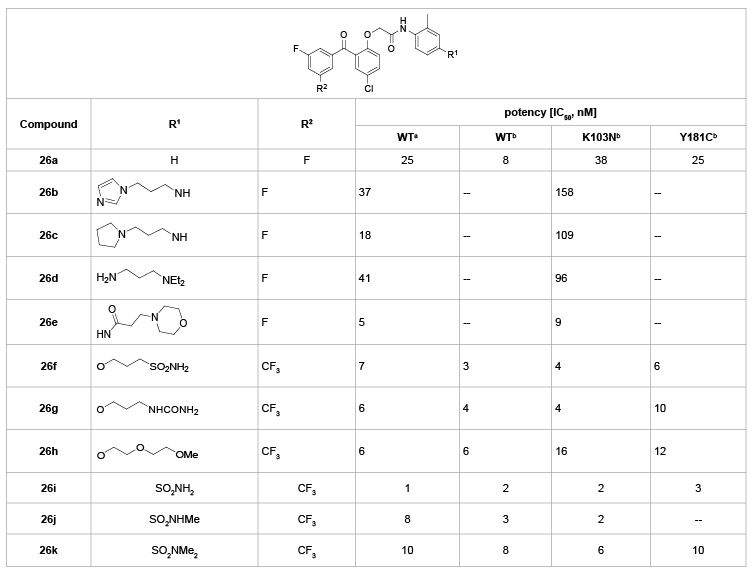
Table 13: SARs of various substituents at the C-4 position of C-ring
a
IC50 values were determined using HIV-1 IIIB wild-type virus in an acute infection assay in MT4 cells; b
IC50 values were determined using a HeLa MAGI
assay system. The wild-type virus was the HxB2 strain, and the K103N and Y181C mutants were isogenic with the HxB2 virus backbone;
c
Undetectable.
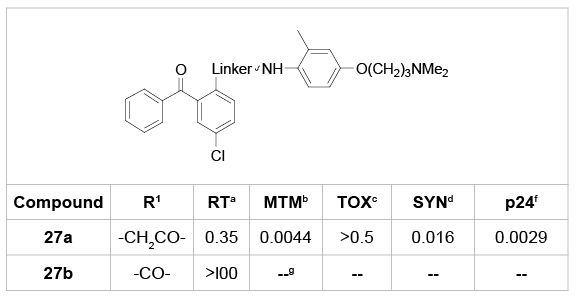
Table 14: SARs of the linker between B- and C-ring.
a
IC50 versus RT of HIV-1 (µg/mL), AZT triphosphate control=0.02 ± 0.005
µM;
b
IC50 versus HIV-1 (strain RF)-infected MT-4 cells (µg/mL), AZT=0.002–
0.02 µM;
c
IC50 versus uninfected MT-4 cells (mg/mL); d
IC50 versus syncytia formation in infected (C8166-cells µg/mL),
AZT=0.002–0.02 µM;
e
IC50 versus p24 antigen formation in infected (C8166-cells µg/mL),
AZT=0.001–0.01 µM;
g
Undetectable.
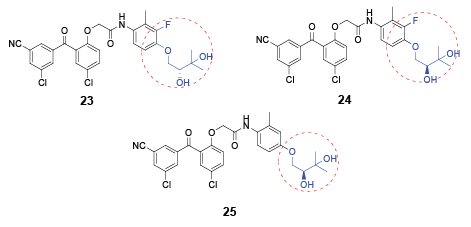
Figure 3: Structures of potent benzophenones with various substituents at C-4 position of C-ring.
In the case of the linker, a very few modifications were performed. In the original paper in 1995, Wyatt et al. [21] reported that reducing the amide linker chain length significantly lost the anti-HIV activity (Table 14). Therefore, more work on modifying this linker is necessary to further explore its SARs.
The use of NNRTIs as an important part of successful combination therapy in HIV/AIDS patients have been well established and widely accepted. In recent years, vast progress has been achieved in the development of NNRTI, especially to improve their antiviral potency. Furthermore, the NNRTI clinical pipeline also seems not to be that impressive as hoped. Nevertheless, the major cause of treatment failure, fast emergence of clinical resistance is still the serious issue for NNRTIs. During the past decade, benzophenone derivatives, have been transformed from a lead with moderate activity against wild-type HIV and little or no activity against clinically relevant mutants to very promising series with excellent in vitro potency against wild-type and key mutant viruses with the unremitting efforts. To provide valuable SAR information for producing more effective benzophenone HIV inhibitors, the SARs of BPs were provided in this manuscript, which were overviewed as below: (1) the carbonyl between the A- and B-ring was necessary for maintaining the pharmacodynamics conformation of this template. (2) Meta substitution on the A-ring would require a polar aprotic and sterically small substituent, such as cyano, methyl, which would reach into the hydrophobic region adjacent to the side chains of Y181 and Y188. (3) The methyl substituent ortho to the amide on the C-ring gave the molecule a measure of metabolic stability, and the para substitution counter the lipophilic nature was much tolerated. In spite of this valuable SAR information, there are still some unclear SARs, for instance, the impact on the activity by changing the linker between B- and C-ring, the impact of the replacement of both Aand B-rings with a bulky aromatic ring, and so on. Therefore, it is essential to extensively optimize this promising template to generate more active anti-HIV agents.
We are grateful to the National Natural Science Foundation of China (No. 81402788), and the PhD Start-up Fund of Natural Science Foundation of Liaoning Province, China (No. 20141115) for the financial support of this research.
Download Provisional PDF Here
Article Type: Review Article
Citation: Ge Y, Xu J, Song Z, Ma X (2016) Benzophenone Derivatives: a Novel Scaffold as Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. J HIV AIDS 2(2): http://dx.doi. org/10.16966/2380-5536.121
Copyright: © 2016 Ge Y, et al. This is an openaccess article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access