Introduction
Erythropoietic protoporphyria (EPP) is a type of porphyria caused by the deficiency of ferrochelatase (FECH; EC4.99.1.1), which is the enzyme involved in the process from protoporphyrin IX to heme in the heme biosynthetic pathway [1]. The first patient with EPP was reported in 1961 by Magnus [2]. The estimated prevalence of EPP is 1:17,000 to 1:200,000 persons [3,4]. Liver dysfunction in patients with EPP began to attract attention after EPP patients with liver cirrhosis were reported for the first time in 1963 [5]. We previously reported a case of siblings with liver dysfunction who had not been diagnosed as having EPP for a long time because of their mild photosensitivity [6]. One sibling died because of liver dysfunction and heart failure. We here report his disease course and liver pathology, which were characteristic and informative in understanding the pathophysiology of the liver damage that occurs in EPP.
Case
The patient was a younger brother of siblings with EPP. There was no family history of liver dysfunction or photosensitivity. He was born at 39 weeks of gestation by normal spontaneous delivery. His older sister was diagnosed before him because of liver dysfunction and photosensitivity. He had a high level of free erythrocyte protoporphyrin (2,779 µg/dL) at the age of 9 years. He did not demonstrate remarkable photosensitivity, and only complained of a photosensitive episode once at 4 years of age. His liver function tests were normal at the time of EPP diagnosis. After his diagnosis with EPP, he was admitted to our hospital owing to abdominal pain several times. Chlorpromazine hydrochloride was administered for his severe abdominal pain, which was effective. Throughout his life, whenever the level of metal-free erythrocyte protoporphyrin decreased to below 3,000 µg/dL, his transaminase level decreased to a nearly normal level, and conversely, an increase in his transaminase level was associated with an increase in protoporphyrin level. From the time of hospital admission, he had been taking ursodeoxycholic acid (URSO: max 900 mg/day) and beta carotene since he was 12 years old, and the level of metal-free erythrocyte protoporphyrin had been between 2,200 to 4,200 µg/dL for several years. His liver function was normal between the time of hospitalization until the age of 28 years (Figure 1). A liver needle biopsy was performed at 23 years of age. Subsequently, his liver dysfunction worsened, which correlated with an increase in protoporphyrin level. His liver function (AST and ALT) was normal between the time of hospitalization until the age of 28 years (Figure 1). At 28 years of age, his liver function rapidly worsened, together with cholestasis. He did not respond to URSO, cimetidine (400 mg/ day), or phlebotomy. He was on the waiting list for a cadaveric liver transplant. In the meantime, he also developed concurrent heart disease with aortic valve regurgitation. We hence planned radical heart surgery; however, heart surgery was not possible because of severe heart failure and liver failure. He subsequently died because of terminal heart failure. Laboratory findings at his terminal stage are shown in table 1. He and his older sister had the c.339delA and IVS3- 48 T/C mutations in the FECH gene. Sequence analysis demonstrated that his father had same heterozygosis 1-base deletion in exon 4 of the FECH gene. The mother did not have any mutations in the same part of exon 4 except IVS3-48 T/C.
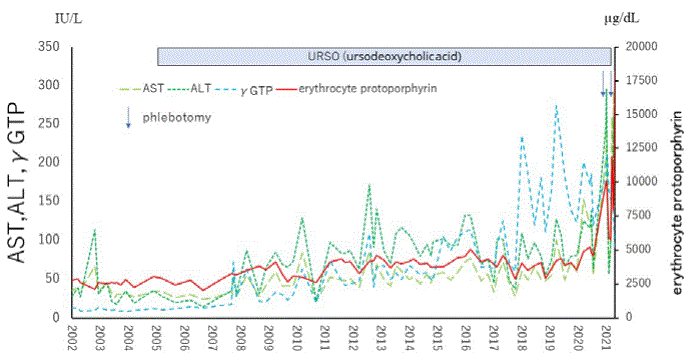
Figure 1: Course of aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transpeptidase (GTP) and metal-free erythrocyte protoporphyrin levels of the patient-Transaminase level increased when the level of erythrocyte protoporphyrin exceeded 3,000 µg/mL.
| |
|
|
normal range |
| WBC |
6,900 |
/µL |
3,500-9,000 |
| Neutrophils |
79.2 |
% |
40-60 |
| Eosinophils |
0.9 |
% |
01-May |
| Basophils |
0.4 |
% |
0-5 |
| Lymphocytes |
14.6 |
% |
18-50 |
| Monocytes |
4.9 |
% |
02-Oct |
| Hb |
10.3 |
g/dL |
13.5-17.6 |
| Ht |
30.1 |
% |
39.8-51.8 |
| Plt |
15.7 |
×104/μL |
15-40 |
| TP |
6.8 |
g/dL |
6.7-8.3 |
| T-Bil |
9.58 |
mg/dL |
0.3-1.2 |
| D-Bil |
7.03 |
mg/dL |
0.0-0.2 |
| AST |
193 |
IU/L |
Aug-33 |
| ALT |
191 |
IU/L |
Apr-45 |
| γ-GTP |
137 |
IU/L |
Oct-47 |
| LDH |
148 |
IU/L |
119-229 |
| ALP |
121 |
IU/L |
38-113 |
| ChE |
99 |
IU/L |
214-466 |
| BUN |
9.4 |
mg/dL |
8.0-22.0 |
| Cr |
0.45 |
mg/dL |
0.6-1.1 |
| amylase |
110 |
IU/L |
37-125 |
| p-amylase |
87 |
IU/L |
21-64 |
| lipase |
89 |
IU/L |
Sep-55 |
| T-chol |
159 |
mg/dL |
128-219 |
| TG |
246 |
mg/dL |
30-149 |
| UA |
2.2 |
mg/dL |
3.6-7.0 |
| Glucose |
104 |
mg/dL |
70-109 |
| Na |
129 |
mEq/L |
138-146 |
| K |
4.5 |
mEq/L |
3.6-4.9 |
| Cl |
94 |
mEq/L |
99-109 |
| NH3 |
59 |
ug/dL |
Dec-66 |
| CRP |
2.01 |
mg/dL |
<0.3 |
| S-Fe |
157 |
µg/dL |
60-200 |
| Zn |
49 |
µg/dL |
80-130 |
| ferritin |
193 |
ng/mL |
39.4-340 |
| Mac-2binding protein |
(2+) |
|
(-) |
| total bile acid |
125.5 |
µmol/L |
<10 |
| hialuronic acid |
592 |
ng/mL |
<50 |
| type 4 collagen 7S |
15.5 |
ng/mL |
<4.4 |
| P-III-p |
42.1 |
ng/mL |
3.62-9.52 |
| BNP |
110.7 |
pg/mL |
<18.4 |
| NT-proBNP |
1,334 |
pg/mL |
<125 |
| |
|
|
|
| Uroporphyrin |
60 |
μg/g・CRE |
<36 |
| Porphobilinogen |
1.2 |
mg/day |
<2.0 |
| gamma-ALA |
0.3 |
mg/ g・CRE |
0.7-2.5 |
| coproporphyrin |
681 |
µg/g・CRE |
<170 |
| RBC protoporphyrin |
9460 |
µg /dL RBC |
<50 |
Table 1: Laboratory data of the patient at 28 years of age.
Autopsy
The patient was lean (height: 184.0 cm; weight: 51.6 kg; BMI: 15.2) with serous atrophy of adipose tissue in the subcutis, omentum, mesentery, and epicardium.
Protoporphyrin was deposited in various organs, such as the liver, spleen, pancreatic head lymph nodes, and para-aortic lymph nodes. His liver was macroscopically hard and dark. Histological analysis revealed cirrhosis with cholestasis, and large amounts of brown pigment in the hepatocytes, Kupffer cells, and bile canaliculi (Figures 2-4). There was congestion in many regions, which was a result of heart failure. Associated lesions included chronic active hepatitis with fibrosis (A2F3), splenomegaly, ascites, pleural effusion, and esophageal varices. Maltese cross findings in polarized light microscopy was visible (Figure 4). Electron microscopic analysis of the liver revealed that the cytoplasmic depositions identified were the same as the crystals described in previous EPP reports as “needle-like crystals” or “starburst patterns” (Figure 4) [7-9].
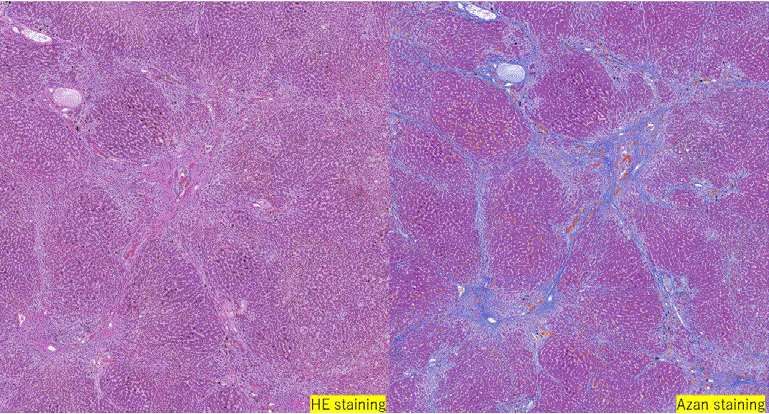
Figure 2: Histopathological findings of the liver at autopsy.
Right: Azan staining (×100). Septum formation and pericellular fibrosis are observed around the central vein.
Left: HE staining (×400) A large amount of brown pigment is observed in hepatocytes and Kupffer cells.
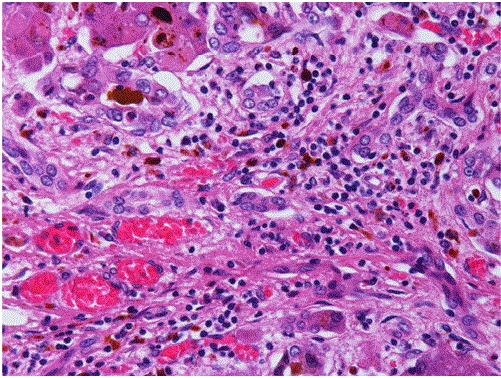
Figure 3: Histopathological findings of the liver at autopsy.
HE staining demonstrates brown pigmentation.
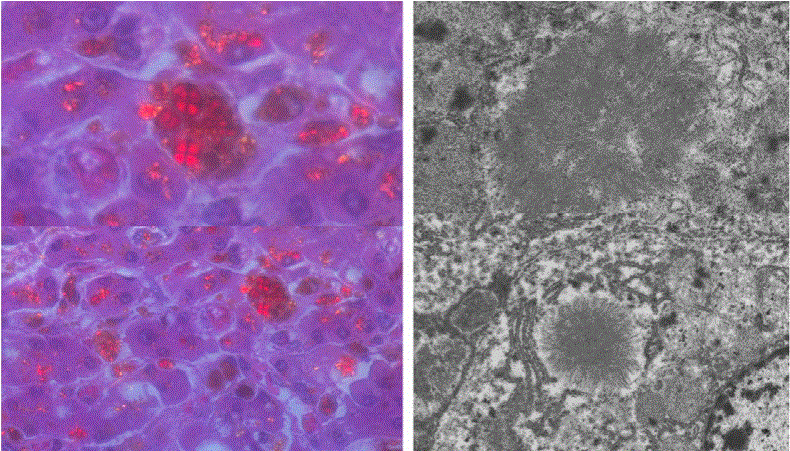
Figure 4: Autopsy findings of the liver.
Polarization light microscopy of liver displayed brown pigmentation like Maltese cross (left), and electron microscopy displayed slender crystals in the cytoplasm (right), which have previously been reported as “needle-like crystals”.
The cardiac lesion showed bicuspid aortic valve (circumference: 9 cm; with fusion of the right coronary cusp and non-coronary cusp) together with fibrotic myxomatous degeneration and thickened valve cusps (Figure 5). The left ventricle was moderately enlarged. There was no evidence of accumulation of protoporphyrin in the heart including valve. He also displayed signs of bronchopneumonia and focal alveolar hemorrhage.
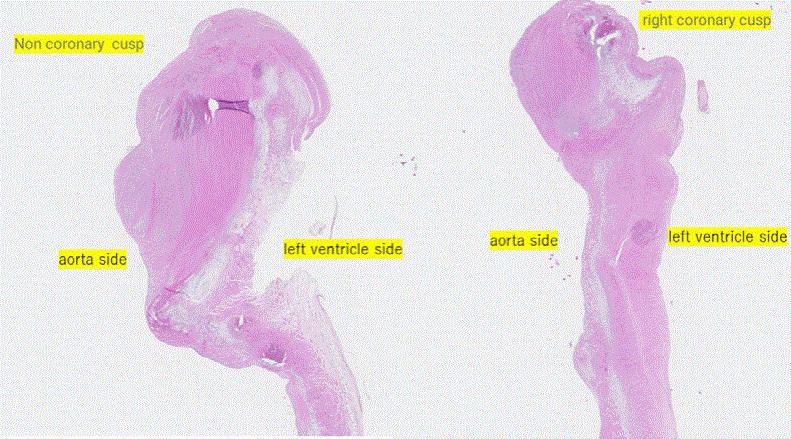
Figure 5: HE staining of the heart at autopsy.
HE staining revealed no characteristic pigmentation of the coronary cusp.
Other findings included acute splenitis, congestion of the spleen, lung, kidney, and liver, and hyperplastic polyps in the gallbladder.
Comparison with his previous liver pathology
A liver needle biopsy had been performed on the patient at 23 years of age because of his liver dysfunction. At that time, his liver was found to be in the pre-cirrhotic state. Azan staining demonstrated that hepatic lobule structures were disrupted, with septum formations and pericellular fibrosis around the central vein. Hematoxylin-eosin (HE) staining showed many brown deposits in hepatocytes and Kupffer cells. In addition, iron staining-positive material-laden macrophages were scattered in the portal region (Figure 6).
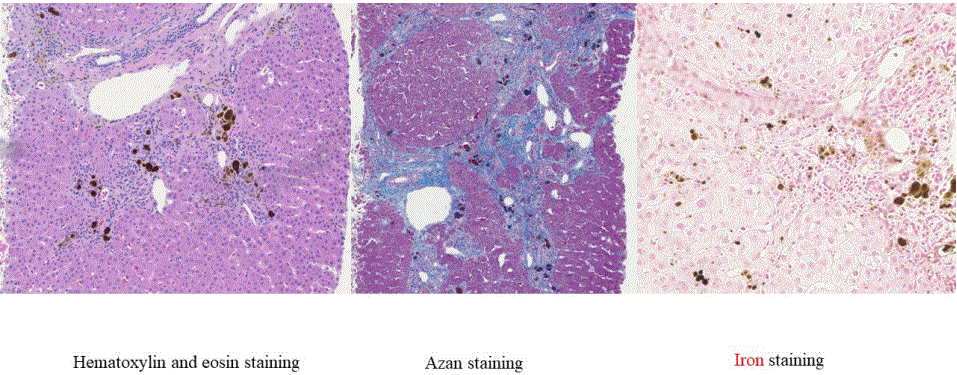
Figure 6: Liver pathology at the age of 23.
Left: HE staining. A large amount of brown pigment is observed in hepatocytes and Kupffer cells.
Middle: Azan staining. Septum formation and pericellular fibrosis are observed around the central vein.
Right: Iron staining. Positive signals are observed in the portal region.
Discussion
In this report, we described a patient with EPP, who showed rapid progression of liver damage. Pathological findings suggested that substantial progression of hepatocellular damage had occurred even though his clinical manifestations (liver function tests) were not so severe. His older sister had spent a long period of time without a correct diagnosis. As she underwent a cardiac operation and transfusion for VSD (Ventricular septal defect) when she was 1 years old, her hepatodysfunction was misdiagnosed as post-transfusion hepatitis until she was 9 years old. The patient (her younger brother) was diagnosed as having EPP owing to his high levels of metal-free erythrocyte protoporphyrin levels and genomic analyses. His liver function tests showed no abnormalities at that time, and he did not complain of photosensitivity. However, at 23 years of age, a liver biopsy was performed because of a mild abnormality in liver function tests. His liver pathology had already shown that his liver was in the precirrhotic stage.
A previous report described that mild abnormalities of liver function may be detected in about 10% of EPP patients. It is estimated that approximately 2 to 5% of EPP patients develop liver failure owing to the toxicity of protoporphyrin on the liver [10,11]. Occasionally, some patients require liver transplantation [12-14]. Without a combined bone marrow and liver transplant, however, the same problems can reoccur and cause long-term problems. Liver damage is initiated by the excess accumulation and subsequent crystallization of protoporphyrin IX within hepatocytes. An excess amount of protoporphyrin in the liver may form solid deposits and cause obstruction of bile flow. Crystalline deposits of protoporphyrin also directly cause cell damage [15].
A large amount of pectinate material was found to exist within the enlarged mitochondria of EPP patients [6]. Rademakers et al. described that they identified giant mitochondria with paracrystalline inclusions in the livers of EPP patients [7], but they considered
that the phenomenon was nonspecific. The general characteristics of liver mitochondria of EPP patients are transformation and the disappearance of cristae [16]. This mitochondrial enlargement may be associated with the involvement of FECH in the heme synthesis pathway within the mitochondria.
Early diagnosis and early disease management are the most important for EPP patients. Regarding unidentified liver dysfunction in patients, erythrocyte protoporphyrin (including total and metalfree fractions) and liver function testing should be monitored from as early as possible, because an increase in either could signal liver dysfunction. Patients should have further work-up for causes of liver dysfunction in the setting of any elevation in liver function testing or a large change in erythrocyte protoporphyrin. If protoporphyria can be diagnosed in patients during childhood, their disease course is expected to be considerably more favorable. In patients with EPP, mutation analysis of the FECH gene may not only confirm the diagnosis, but may also suggest an increased risk of liver disease. EPP patients with a null variant of FECH along with IVD-48T>C, or with two rare pathogenic variants appear to have an increased risk of liver disease [17-19]. The patient in this study and his sister had the IVD48T>C and c.339delA variants, which increase the risk of liver disease.
In this study, the siblings both had heart disease. The older sister was found to have ASD (Atrial Septal Defect) and VSD during the neonatal period, and underwent radical surgery when she was 8 months old. However, she and their relatives had no heart valve problems. Therefore, the heart valve problem of the patient was unlikely to be genetic. There are only 3 reports of EPP patients with heart disease. Therefore, it is a very rare case. However, in a bovine model of EPP, average FECH activity was decreased to about one-tenth of normal in the heart and lung [20]. Heme has a negative effect on the heart, but the role of protoporphyrin IX on the heart has not been clarified to date.
Yotsumoto, et al. reported a 52-year-old man with EPP who underwent aortic valve replacement owing to severe regurgitation. To prevent burn injuries, the astral lamps in the operating room were covered with yellow film filters. Blood priming of the extracorporeal circuit was performed to maintain adequate hemoglobin concentrations, which resulted in the reduction of heme synthesis. The patient was discharged in good health without any adverse events from EPP. However, this patient did not have liver dysfunction [21].
Another paper reported a 58-year-old male patient with EPP who underwent aortic valve replacement and a concomitant aortocoronary bypass. The patient has been followed without complications of EPP postoperatively. Therefore, cardiac surgery can safely be performed on patients with EPP [22].
During heart failure, ischemic liver injury occurs due to decreased hepatic blood flow and hypoxia. In addition, hepatic congestion occurs when hepatic venous return is impaired owing to congestive heart failure. Liver damage caused by hepatic congestion is called congestive hepatopathy, and the chronic persistence of this condition leads to congestive liver cirrhosis. Chronic hepatic congestion causes liver dysfunction and liver fibrosis, leading to congestive cirrhosis. It is known that acute ischemic lesions overlap with chronic congestive liver injury, resulting in the so-called acute on chronic (acute exacerbation) condition, which can become severe [23,24].
Free heme can be toxic as it catalyzes the production of reactive oxygen species, oxidizes lipids and proteins, and causes DNA damage, thereby inducing a proinflammatory environment. Extensive research has clarified the underlying mechanisms by which heme contributes to the development of cardiovascular diseases through oxidative stress, gene expression regulation, and phenotypic changes in cells. Excess heme plays a detrimental role in atherosclerosis, heart failure, myocardial ischemia-reperfusion injury, degenerative aortic valve stenosis, and cardiac iron overload. Thus, heme concentrations beyond normal levels are dangerous [25]. Our present patient demonstrated that liver damage in EPP can progress rapidly, particularly when patients have heart disease.
Acknowledgments
Ethics approval and Consent to participate
This case report was written in accordance with the Declaration of Helsinki. Consent to publish: Written informed consent was obtained from the patient and his parents regarding presentation of the case, including its publication, with proper consideration of the patient’s personal information. Competing interests: The authors have no competing interests to declare. Funding: No external funding was obtained for this study.
Author contributions:
N.S. and H.K. designed the study; C.M., N.R., and Y.K. collected the data and N.S. wrote the manuscript; T.K., K.M., and N.T. provided technical support and conceptual advice. All authors read and approved the final manuscript. H.K. critically reviewed the manuscript and supervised the whole study process. All authors read and approved the final manuscript.
Article Information
Article Type: CASE REPORT
Citation: Nishimata S, Chiyotanda M, Nagao R, Tasaki K, Kajiwara M, et al. (2023) Case of Rapid Progression of Liver Damage in a Patient with Erythropoietic Protoporphyria Accompanied with Bicuspid Aortic Valve. Int J Endocrinol Metab Disord 8(1): dx.doi.org/10.16966/2380- 548X.177
Copyright: © 2023 Nishimata S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 17 Jan, 2023
Accepted date: 16 Feb, 2023
Published date: 23 Feb, 2023
