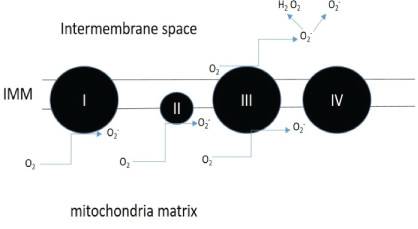
Figure 1: Sites of superoxide production.
Complex I to III of the electron transport chain are main producers of the superoxide which is converted to hydrogen peroxide by superoxide dismutase enzymes found within the mitochondria [16].

Ali Maria1 Aghadi Maryam2 Vincent Annette Shoba1*
1Biological Sciences Program, Carnegie Mellon University, Qatar*Corresponding authors: Vincent Annette Shoba, Biological Sciences Program, Carnegie Mellon University in Qatar, Education City, PO box 24866 Doha, Qatar, Tel: +974 44848523; E-mail: annettev@qatar.cmu.edu
Aspartame is a low calorie sugar that is widely used in artificial sweeteners. Although different studies indicated associated health symptoms to it, it continues to be controversial. Studies have also shown consumption of aspartame caused neurological deficits and mitochondrial mediated activation of apoptosis was observed as a long-term effect of aspartame in rat brain. In this study, the biochemical responses of distal tubular kidney cells upon short-term exposure to aspartame were measured. MDCK Type II (kidney) cells were exposed to 50 µg/ml to 250 µg/ml aspartame for 30 mins. Mitochondrial linked biochemical responses such as oxidative stress and energy production was measured. Overall increase in dehydrogenase activity together with increased production of ATP was observed. This was accompanied by an overall increase in mitochondria derived oxidative stress. However, mitochondrial membrane potential and cellular NAD/NADH ratio in the aspartame exposed cells remained unchanged. These results indicate that while aspartame did not cause significant changes in the mitochondrial activity, the elevated activity of the mitochondria shown through the increase in ATP production and oxidative stress indicate the involvement of the mitochondria in aspartame-mediated cellular response. More studies would need to be done to clarify the mechanism by which aspartame increases oxidative stress via the mitochondria.
Oxidative stress; Mitochondria; Aspartame; Malate-aspartate shuttle
MDCK-Madin-Darby Canine Kidney; ATP-Adenosine Triphosphate; NAD+- Nicotinamide Adenine Dinucleotide; NADHreduced Nicotinamide Adenine Dinucleotide; •O2--Superoxide Anion; H2O2 -Hydrogen Peroxide; AOA-Aminooxyacetate; DCF-HAD-2′,7′- Dichlorodihydrofluorescein diacetate; MTT-3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; MAS-Malate Aspartate Shuttle; MMP or ΔΨm- Mitochondrial Membrane Potential; IMM-Inner Mitochondrial Membrane; JC-1-1H-Benzimidazolium, 5,6-dichloro-2-[3-(5,6-dichloro-1,3- diethyl-1,3-dihydro-2Hbenzimidazol-2-ylidene)-1-propenyl]-1,3-diethyl-, iodide
Artificial sweeteners are food additives that are used as sugar substitutes. They have become extremely popular over the past couple of years as they claim to promote weight loss and are considered safe for consumption by diabetics. Artificial sweeteners such as aspartame, saccharin and stevia have been recognized as safe for consumption by the United States Food and Drug Administration (FDA). The major benefits of artificial sweeteners are that these chemicals do not have an influence on plasma glucose or insulin level, hence their use in different soft drinks and foods [1]. Aspartame, therefore, was widely used by diabetic and obese people as it helped reduce energy intake and modulate blood glucose levels. However, many health concerns have been raised regarding the toxicity of aspartame. Aspartame metabolism was shown to generate methanol as a by-product, which was responsible for oxidative stress in rats [2], while, other research suggest that aspartame fed rats developed brain tumors [3]. In a study by Horio Y, et al, aspartame induced cell death in PC12, a neuronal cell line, was suggested to involve the mitochondrial pathway of apoptosis through the generation of reactive oxygen species (ROS) [4]. An epidemiological study also indicated that aspartame consumption had the potential to cause cancer development. The study compared 197 patients diagnosed with urinary tract tumors with 397 controls. The authors reported that the risk of urinary tract tumors was significantly increased upon long-term exposure (more than 10 years) to artificial sweeteners, including aspartame [5]. Besides urinary tract tumors, the carcinogenic effect of aspartame was also reported [6]. In albino rat models, daily administration of aspartame resulted in reduced activities of antioxidant enzymes and increased oxidative stress markers in the kidney and liver [7].
Oxidative stress is the lack of balance between the production of ROS and antioxidant defenses [8], which leads to a state where oxygen free radicals exceed the antioxidant systems of the cells [9]. The free radicals: oxidants or ROS such as •O2 - (superoxide) and H2O2 (hydrogen peroxide) are usually generated by the mitochondria in a normal cellular metabolism and are eliminated by antioxidants such as ubiquinol-10 vitamin C, and E [7]. Mitochondria are the major production site of ROS whereby electrons leak out from the electron transport chain and combine with molecular oxygen to form •O2 - , a precursor of most ROS. Sites of superoxide formation are depicted in Figure 1. The contribution of each site to the overall superoxide level is organ specific and also depends on how reduced the respiration chain is [10-12]. Higher superoxide production is anticipated when the mitochondria are not synthesizing ATP and have a high mitochondrial membrane potential due to the sluggish electron transfer. The imbalance due to oxidative stress can damage important biomolecules such as the DNA or lead to lipid peroxidation affecting the cell membrane and the cell as a whole [9]. The sustenance of this oxidative stress can cause production of ROS over a long time which may lead to somatic mutations and neoplastic transformation [8]. Consequently, increased DNA mutations or damage can cause abnormal cell proliferation and the formation of cancers [8]. One study specifically examined the effect of aspartame on Madin-Darby Canine Kidney (MDCK) cells. Here, a decrease in cell viability was found on exposure to aspartame as well as an increase in mitochondria derived reactive oxidative species in a dose dependent manner. This study suggested that aspartame treatment may cause oxidative stress in MDCK cells [13].
Figure 1: Sites of superoxide production.
Complex I to III of the electron transport chain are main producers of the superoxide which is converted to hydrogen peroxide by superoxide dismutase enzymes found within the mitochondria [16].
The hydrolysis of aspartame into its amino acid components: aspartate and phenylalanine, is hypothesized in this study, to increase intracellular oxidative stress through the malate aspartate shuttle. Under normal conditions, the cycle begins with the conversion of aspartate to oxaloacetate, which is then reduced to malate. Malate can enter the mitochondrial matrix, where it is oxidized back to oxaloacetate, which is converted to aspartate and transported back into the cytosol, where the cycle begins again [14]. The electron gained in the oxidation of malate to oxaloacetate enters the electron transport chain, which produces reactive oxidative species. If the amount of aspartate is increased, the activity of the malate aspartate shuttle will increase as well [14].
In this study, the main objective was to elucidate the mechanism of the acute effect of aspartame on oxidative stress in Madin-Darby Canine kidney cells. This was carried out by measuring mitochondrial function through measurements of the activity of the malate aspartate shuttle, intracellular ATP production, changes in mitochondrial membrane potential and quantitation of mitochondrial derived oxidative stress. Elevated activity of mitochondrial dependent processes such as the citric acid cycle could thereby producing large amounts of reactive oxidative species and causing an increase in oxidative stress [15].
Madin-Darby Canine Kidney (MDCK) Type II cells were purchased from the European Collection of Authenticated Cell Cultures (ECACC). Aspartame, ATP bioluminescent somatic cell assay kit (FLASC), reduced β-Nicotinamide adenine dinucleotide (NADH) reduced disodium salt, aminooxyacetate (AOA), 2′,7′-Dichlorodihydrofluorescein diacetate (DCF-HDA), Trizma Base, Tris-HCl, aspartame, phosphate buffered saline (PBS), Dulbecco’s Modified Eagle’s medium (DMEM) were obtained from Sigma-Aldrich (St. Louis, MO, USA). NAD/NADH Colorimetric Assay Kit was purchased from Abcam Inc. (Cambridge, MA). Mitosox Red, Trypsin, fetal bovine serum (FBS), and penicillin streptomycin were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Madin-Darby Canine Kidney (MDCK) Type II cells were maintained in DMEM supplemented with 10% FBS and 1% penicillinstreptomycin in a humidified incubator at 37° C with 5% carbon dioxide. The cells were allowed to adhere overnight in various culture vessels and the medium was then replaced with culture medium without or with various concentrations of aspartame.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay: MDCK cells were plated at a density of 10,000 cells per well in a 96-well plate and left to adhere overnight at 37° C with 5% carbon dioxide. The following day, cells were exposed to aspartame prepared in culture media at concentrations ranging from 0 to 250 μg/ ml at 37° C for 0.5 and 1 hour. This was followed by incubation of cells in 0.5 mg/ml MTT, prepared in cell culture medium, for 2 hours. The reduced purple formazan product was solubilized in 50 μl dimethyl sulfoxide (DMSO) and quantification of the reduced formazan was carried out using the TECAN Sunrise microplate reader at 550 nm. Triplicates were performed for all samples.
Mitochondrial membrane potential: The BD MitoScreen (JC-1) kit was used to prepare cell samples for measurement through flow cytometry. Firstly, MDCK cells in T 25 flasks were treated with 50 μg/ mL or 250 μg/mL of aspartame for 30 minutes in a CO2 incubator. The cells were trypsinized and centrifuged at 1000 rpm for 5 min. The cells were resuspended in 0.2 μM JC-1 working solution (prepared using the protocol provided with the kit) and incubated in the CO2 incubator for 15 minutes. 2 mL of 1X Assay Buffer was added to all cell samples. The samples were centrifuged at 400 × g for 5 min and resuspended in 1 mL of 1X Assay Buffer. The samples were centrifuged once more under the same conditions and resuspended in 1X Assay Buffer. The cells were analyzed using the BD LSR Fortessa. Average fluorescence readings were taken at 530 nm and 590 nm. Triplicates were performed for all samples. For fluorescence imaging, 50,000 MDCK cells were placed on a 6 well plate and left to adhere overnight in a CO2 incubator. The next day, the cells were treated with 0.2 μM JC-1 (BD Biosciences) for 15 minutes. Images for the cells were taken using the EVOS fluorescence microscope (Life Technologies) under the GFP and RFP filters. Next, the JC-1 solution was removed from the wells and PBS, 50 μg/mL and 250 μg/mL were introduced into one of each well. Images for the wells were taken right after adding the treatment and also at the end of 30 minute incubation with the treatments in a CO2 incubator.
Intracellular ATP levels: 50,000 MDCK cells were placed on a 24- well plate and left to adhere overnight at 37° C with 5% carbon dioxide. The following day, cells were washed with PBS and exposed to varying concentrations of aspartame ranging from 0 to 250 μg/ml at 37° C for 0.5 and 1 hour. Cells were washed with PBS and lysed with ice-cold water. Using a pipette tip, cells were scrapped off the wells and the lysates were boiled for 3 mins following which ATP determination was carried out using the procedure provided in the Sigma FL-ASC Bioluminescent somatic cell assay kit (Sigma-Aldrich). Measurements of the chemiluminescence of the luciferin-luciferase reaction were made using a 96-well plate and read in a luminometer (Victor3, PerkinElmer). The amount of ATP in each sample was extrapolated from a standard curve generated from 2 to 100 pmol ATP, a range that produced a linear response. Intracellular NAD+/NADH ratio NAD+ and NADH levels were determined separately using a NAD/NADH Assay Kit (Colorimetric) (Abcam Inc.). Briefly, in order to measure total levels of NAD+ and NADH, lysates obtained from MDCK cells exposed to aspartame for 30min were transferred into a 96-well plate. Measurements were done as described in kit. A standard curve was generated by serial dilution of standard NADH solution and the NAD+/NADH ratio was calculated as [(NADtotal- NADH)/NADH].
C57BL/6NJ mouse strain was euthanized by exposing them to CO2 for 15 mins. Kidney tissues obtained from them were washed and homogenized in ice cold isolation buffer (2 mM Tris-HCl, 250 mM sucrose, 4 mM potassium chloride, 2 mM EDTA, and 04 g/L BSA at pH 7.5). The extracted kidneys were placed in 20 mL of the isolation buffer and cut into small pieces. The suspension was placed in a glass homogenizer and the kidneys were further ground into finer particles using a tight fitting glass pestle. The homogenate was transferred to a 50 mL tube and centrifuged at 1000 × g for 10 minutes in the Sorvall ST 16 Centrifuge (Thermo Fisher Scientific). The supernatant was extracted and centrifuged again at 10,000 × g for 10 minutes. The supernatant was discarded and the pellet was resuspended in 1 mL of the isolation buffer. The resulting volume was diluted 4 times and centrifuged again at 10,000 × g for 10 min. The resulting pellet containing mitochondria was resuspended in 300 μL of the isolation buffer. The suspension was kept on ice when measuring Malate Aspartate Shuttle (MAS) activity. Bradford Assay was performed to determine protein concentration of the mitochondrial suspension.
Malate aspartate shuttle (MAS) activity: On the same day as the isolation of mice kidney mitochondria was performed, malate aspartate shuttle activity was measured for the mitochondria. Measurement of the malate aspartate shuttle activity for the control was performed as such: 2 mL of a buffer containing 300 mM mannitol, 10 mM KH2PO4, 10 mM Tris-HCl, 10 mM KCl, 5 mM MgCl2, and 2 mM aspartate at pH 7.4 was mixed with 2 mM ADP, 0.035 mM NADH, 3 U/mL MDH and 2 U/mL aspartate transaminase (AST) in a quartz cuvette. 0.1 mg of protein from the mitochondrial suspension was added to the cuvette and baseline oxidation of NADH was measured at 340 nm for 2 min using the Agilent Technologies Cary 8454 UV-Vis. Once the baseline oxidation rate became constant, 5 mM glutamate and 5 mM malate were added to the cuvette to initiate malate aspartate shuttle activity, which was measured for the next 4 minutes. For the negative control, 0.2 mM AOA was introduced with the mitochondria extract and MAS activity was measured as described above. For aspartame treatments, mitochondria extract was incubated with 50 μg/mL aspartame, 250 μg/mL aspartame, 50 μg/mL aspartame with 0.2 mM AOA and 250 μg/ mL aspartame with 0.2 mM AOA for 30 minutes. The MAS activities for these samples were measured as described for the control, and the buffer used here lacked the 2 mM aspartate. Triplicates were performed for all samples.
ROS generation assay: ROS generation was measured using flow cytometry. Equal numbers of cells were seeded into T25 flasks and left to adhere overnight at 37° C with 5% carbon dioxide. The next day, cells were washed and exposed to 100 μM DCF-HDA for 30min. Cells were then treated with varying concentrations of aspartame ranging from 0 to 250 μg/ml prepared in HBSS/glucose for 30 min at 37°C. The cells were trypsinized and centrifuged at 1000 rpm for 5 min. The cell pellets were resuspended in 300 μl HBSS/glucose and was analyzed using the BD LSR Fortessa (BD Biosciences) using the Fluorescein Isothiocyanate (FITC) channel Ex/Em 495 nm/519 nm. Triplicates were performed for all samples.
Mitochondrial superoxide production using MitoSox Red: MDCK cells in T25 flasks were incubated with 5 μM Mitosox Red for 10 minutes at 37° C. The cells were then treated with 50 μg/mL or 250 μg/mL of aspartame for 30 minutes. After the treatment, the cells were washed with PBS and trypsinized. The cells were resuspended in PBS and analyzed using the BD LSR Fortessa (BD Biosciences). Average fluorescence readings for the cells were taken at 590 nm. Triplicates were performed for all samples.
Statistical analysis: The significance of the differences between aspartame-treated and respective controls was determined using the Student’s t-test. Values were expressed as the mean ± standard deviation (SD), and were calculated from three independent experiments. A value of P<0.05 was considered statistically significant, and represented by asterisks: *, P<0.05.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) is a tetrazolium salt that is reduced in by dehydrogenases or other enzyme systems where redox equivalents are produced. Therefore, the reduction of MTT is used as a measure of cell viability as well as to indicate the activities of dehydrogenases within the cells. MDCK cells exposed to varying concentrations of aspartame (10 μg/ml to 250 μg/ml) showed no changes in dehydrogenase activity at 30 min indicating no compromise in cell viability (Figure 2). However, upon incubation for 1h, cells showed significantly elevated dehydrogenase activities, compared to controls, at concentrations 20, 50 and 100 μg/ml. This elevation was not significant at higher concentrations of aspartame (Figure 1). These results indicated the possible stimulation of dehydrogenase activities within the cell by aspartame, likely through its by-product aspartate.
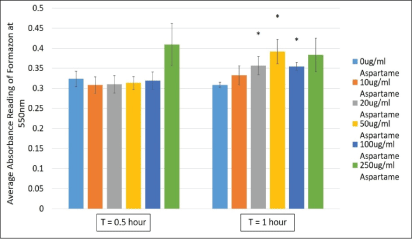
Figure 2: Cell viability measured in MDCK cells treated with different concentrations of aspartame for 30 minutes and 1 h.
MDCK cells were treated with aspartame for 30 min and 1 h. Cells were exposed to 3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and absorbance values of the solubilized formazan product was measured at 550 nm. Values are considered significant if *p value <0.05 for n=3.
Aspartame is composed of phenylalanine and aspartic acid (aspartate) linked together by methanol. The body breaks down aspartame into its by-products. The involvement of aspartate as an anaplerotic carbon source of the TCA cycle through fumarate and oxaloacetate; led us to test the effect of aspartame on the mitochondrial membrane potential (MMP) which is generated through the electron flux on the inner mitochondrial membrane (IMM), contributed by NADH and FADH2. These electron carriers are generated at the TCA cycle for the purpose of transferring the electrons to the electron transport chain on the IMM.
JC1 is a dye that enters the mitochondria as monomers that emit fluorescence in the green wavelength. The presence of mitochondrial depolarization (membrane potential) results in the aggregation of these monomers to give rise to J-aggregates that then emit fluorescence in the red wavelength. The cells were analyzed using flow cytometry and fluorescence microscopy and the results are represented in figures 3A and B respectively. Figure 4 shows that there is an increase in the ratio of red to green fluorescence for cells treated with 50 μg/mL aspartame and that there is a slight decrease in the ratio value for cells treated with 250 μg/mL aspartame as compared to the control as assessed by flow cytometry. However, none of these differences were significant. Figure 5 shows the images of MDCK cells before and 30 minutes after their respective treatments showed clear red fluorescence indicating energized mitochondria in both aspartame treated and non-treated MDCK cells.
Despite ∆Ψm being unaffected, significantly higher levels of ATP was generated in cells that were treated with 250 μg/ml of aspartame within 30 min of exposure as shown in figure 4. The malate-aspartate shuttle involves the glutamate-aspartate carrier and the 2-oxoglutarate (α-ketoglutarate) carrier exchanging malate for 2-oxoglutarate. The shuttle serves to transport of cytosolic NADH into the mitochondrial matrix. NADH is utilized for respiration which generates ATP and regulation of cytosolic levels of NADH (increase due to increased levels of aspartate in the cytosol) contributes to the elevated levels of ATP in the cells. Cytosolic NADH levels were found to be regulated by the glutamate-aspartate carrier during glucose-stimulated insulin secretion in beta-cells [16]. Therefore, the NAD+/NADH ratio in the aspartame treated cells were tested.
NAD is a dinucleotide made up of with one nucleotide containing an adenosine ring, and the other containing nicotinamide. In cellular metabolic pathways, NAD is involved in redox reactions, carrying electrons from one reaction to another. As NAD, it functions as an oxidizing agent where it receives electrons and forms NADH, which in turn can function as a reducing agent to donate electrons. NAD+/ NADH concentrations measured for untreated and cells treated with 0.2 mM AOA served as controls for the experiment. AOA is a known inhibitor of the malate aspartate shuttle, and since the shuttle activity is being inhibited in the AOA treated cells, there will be a decrease in NAD+/NADH ratio. As shown in figure 5, there were no differences in the NAD+/NADH ratios observed in cells with or without aspartame after 30 min.
The malate aspartate shuttle activity was expected to be significantly lower for mitochondria treated with 0.2 mM Aminooxyacetate (AOA) as it is a known inhibitor of the shuttle. Even though figure 6 showed decrease in activity is observed for mitochondria treated with AOA+250 μg/mL aspartame and AOA+50 μg/mL aspartame, they are not significant. Moreover, the differences in activity for mitochondria treated with 250 μg/mL and 50 μg/mL aspartame are not significant. This is most likely due to varying rates of MAS activity among triplicates within specific treatments. However, overall MAS remained constant despite the presence of aspartame. This could be due to the lack of the whole cellular system comprising of the cytosolic and mitochondrial parts of the cell working in unison as the MAS activity was measured in isolated kidney mitochondria.
DCF-HDA assay data is represented in figure 7 where oxidative stress was assessed for 30 minutes of exposure to different aspartame concentrations. The theory behind this assay is that non fluorescent fluorescein will fluoresce when oxidized by reactive oxygen species, consequently, when applied to intact cells DCFH-DA crosses the cell membrane and is hydrolyzed by esterases to DCFH, that is then oxidized by reactive oxygen species to DCF (dichlorofluorescein) that is highly fluorescent [12]. MDCK cells exposed to 250 μg/ml aspartame showed significantly higher oxidative stress compared to the control after 30 min of exposure. However, lower concentrations of aspartame did not show such stress.
MitoSO™ Red indicator is a derivative of dihydroethidium which is a probe that is taken up into actively respiring mitochondria. The Oxidation of MitoSOX™ Red indicator (or HE) by superoxide results in hydroxylation at the 2-position. 2-hydroxyethidium (and the corresponding derivative of MitoSOX™ Red indicator) exhibits a fluorescence excitation peak at around 400 nm that is absent in the excitation spectrum of the ethidium oxidation product generated by ROS other than superoxide. Thus, fluorescence excitation at 400 nm with emission detection at around 590 nm provides optimum discrimination of superoxide from other ROS. The graph in figure 8 shows that there is a significant increase in superoxide production for cells treated with 250 μg/mL aspartame for 30 minutes (170 RFU) as compared to the control (153 RFU). This suggests that cells treated with 250 μg/mL aspartame have elevated superoxide production in the mitochondria as compared to normal MDCK cells.
Over the past years, the human diet has been increasingly exposed to artificial sweeteners as it is considered as healthier alternatives for consumers who want to reduce their sugar intake [17].
Aspartame is one of the most commonly used artificial sweeteners. It was given approval for marketing by the FDA in 1981 and is now used as a sugar replacement in cereals, topping mixes, frostings, confections, chewing gum, dry mix beverages, and yogurt [15]. It is a synthetic combination of the two naturally occurring amino acids aspartic acid and phenylalanine which allows it to be digested like a protein in the body and has a caloric value of 4 kcal/mg, despite the fact that it is 180-200 times sweeter than sucrose [15]. Despite being the most rigorously ever tested food ingredient [16], different studies reporting on the health issues associated with aspartame consumption continues to cast a controversial light on the use of this sweetener.
Studies on animal models have shown consumption of aspartame leading to glucose intolerance and obesity [17], cancer [18,19] and liver dysfunction [16] and nephrotoxic effects [20].
Experimental studies of the toxicological effect aspartame in both animal and cell models have been linked to oxidative stress. Brains of rats exposed to aspartame long-term showed altered antioxidant status indicating free radical generation [2]. MDCK cells showed significant decrease in cell viability and activity of enzymes associated with ROS in cells, namely, catalase and superoxide dismutase [11]. Levels of lipid peroxidation in these cells significantly increased [11]. However, this study measures these effects after 24 to 72 hours of exposure. In our study, the acute effect of aspartame exposure of MDCK cells was evaluated and the possible biochemical mechanism of aspartame toxicity was elucidated.
Short term exposure (30 min) of aspartame to MDCK cells at concentrations between 20 to 100 μg/ml showed no effect on cell viability, however, following 1h exposure cells exhibited increased formazan products which is representative of increased dehydrogenase activities (Figure 2). Dehydrogenases in cells are mainly located within the mitochondria. In figure 3A, changes to energization of the mitochondria within the MDCK cells exposed to aspartame for 30 min showed no significant changes. The ratio of green monomers to red-aggregates in both the control and both low and high concentrations of aspartame remained similar, indicating the mitochondrial membrane potential (∆Ψm) of the aspartame treated cells did not change significantly from the untreated control. The ∆Ψm is highly dependent on the FoF1-ATPase whose activity maintains homeostatic ∆Ψm unless uncoupling of the membrane occurs; this regulation is seen even during H2 O2 -induced oxidative stress [11]. Therefore, a change to the ∆Ψm was not expected. Despite aspartame not affecting ∆Ψm, intracellular ATP production by aspartame treated MDCK cells showed significant increase (Figure 4) compared to the untreated control at concentrations at and above 50 μg/ml. This could be explained by the elevated dehydrogenase activity observed in the treated cells (Figure 1) following 1 hour exposure. The production of NADH by the dehydrogenases feeds into the electron transport chain to generate ATP in cells. The involvements of the shuttle systems on the mitochondrial membrane, namely, the malate-aspartate shuttle (MAS), transport NADH into the mitochondria. Therefore, alongside measuring the NAD+/NADH ratio in treated cells, the MAS activity in isolated mice kidney mitochondria was measured. Both NAD+/ NADH ratio and MAS activity remained unaffected (Figures 5 and 6) by aspartame treatment. However, the absence of the whole cellular system in when measuring MAS activity could have caused the inability to see elevated activity upon aspartame treatment.
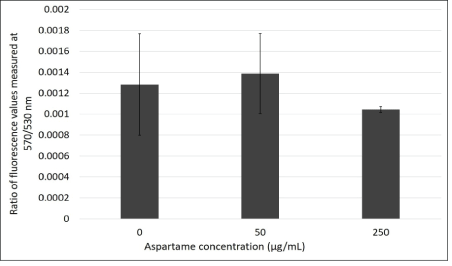
Figure 3A: Ratio of absorbance values measured at 570 nm/530 nm through JC-1 flow cytometry in the presence of aspartame treatment in MDCK cells.
MDCK cells were treated with JC-1 dye and their respective treatments. Absorbance values were measured for each type of treated cells using flow cytometry. Ratios of absorbance values at 570 nm/530 nm were plotted to determine difference in mitochondrial membrane potential for the three different types of cells treatments. Values are considered significant if p value <0.05 for n=3.
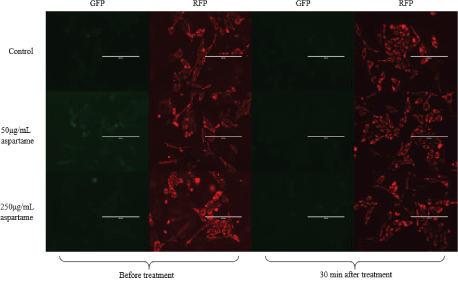
Figure 3B: Fluorescence microscopy images of aspartame treated MDCK cells.
Images were taken of aspartame treated MDCK cells in 6 well plates using the EVOS fluorescence microscope (Life Technologies). Images were taken using the two filters for RFP and GFP.
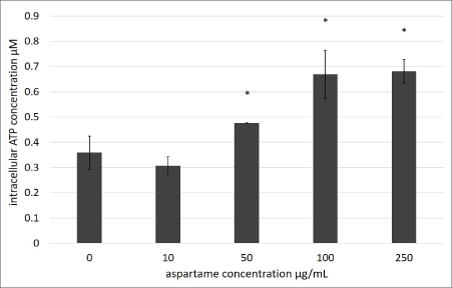
Figure 4: Intracellular ATP production of aspartame treated MDCK cells.
Intracellular ATP of MDCK cells treated with varying concentrations of aspartame for 1 h was determined using the Sigma FL-ASC Bioluminescent Somatic cell assay kit (Sigma-Aldrich). Values are considered significant if *p value <0.05 for n=3.
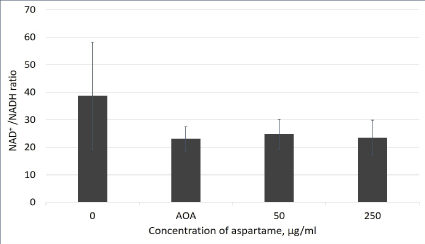
Figure 5: NAD+/NADH ratio in aspartame treated MDCK cells.
Cells indicated by the aspartame bar were treated with 50 and 250 μg/mL of aspartame for 30 minutes. Cells indicated by the AOA bar were treated with 0.2 mM AOA for 10 minutes. Control cells were left untreated. Intracellular NAD/NADH ratios were determined using the NAD/NADH Assay Kit (Colorimetric) (Abcam). Values are considered significant if p value <0.05 for n=3.
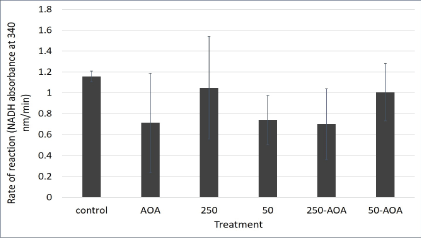
Figure 6: Malate aspartate shuttle activity in aspartame treated mitochondria derived from mouse kidney.
Mitochondria indicated by the 250 bar were treated with 250 μg/ mL of aspartame. Mitochondria indicated by the 50 bar were treated with 50 μg/mL of aspartame. Mitochondria indicated by the bars 50- AOA and 250-AOA were treated with the indicated concentration of aspartame as well as 0.2 mM AOA. All treatments were performed for 30 minutes. Values are considered significant if p value <0.05.
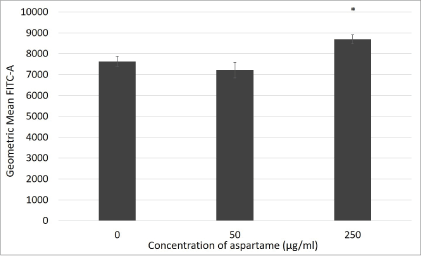
Figure 7: Reactive oxygen species production in aspartame treated MDCK cells.
Cells were incubated with their various treatments for 30 minutes. Reactive oxygen species production was measured through flow cytometry using DCF-HDA probe. Fluoresence are expressed as geometric mean of green fluorescence. Values are considered significant if *p value <0.05 for n=3.
The findings in previous studies show increased ROS in cells following longer exposure times to aspartame [13], however we find increased ROS within 30min of exposure of MDCK cells to aspartame at the concentration of 250 μg/ml. The contribution of the mitochondria to the production of the oxidative stress, namely superoxide production, was clearly elucidated in Figure 8 where MDCK cells exposed to 250 μg/ml aspartame showed significant increase in mitochondrial superoxide production within 30 min of exposure compared to the untreated control. One interesting conclusion to these results is the likely involvement of 2-oxoglutarate dehydrogenase. In a report by Starkov AA, et al. [21], in mitochondria isolated from mouse brain 2-oxoglutarate dehydrogenase was found to be the primary site of ROS production in normally functioning mitochondria [21,22]. We hypothesize that over active 2-oxoglutarate dehydrogenase due to elevated aspartate levels was the main reason to elevated ROS and superoxide levels in the MDCK cells.

Figure 8: Superoxide production in mitochondria of aspartame treated MDCK cells.
Cells were incubated with their various treatments for 30 minutes. Superoxide production was measured through Mitosox red flow cytometry. Fluorescence values for cells were measured at Ex/Em 510 nm/590 nm. Values are considered significant if *p value <0.05 for n=3.
In addition, in an in sillico model reported by Lu M, et al. [23] the 2-oxoglutarate dehydrogenase complex binds both aspartate aminotransferase and malate dehydrogenase to form a ternary complex that can enhance malate oxidation in the presence of malate and glutamate [23]. Such complexes can effectively increase the reaction rates by maintaining the TCA cycle intermediates concentrations and reduce the transit time of intermediates between enzymes Further to this, it also enhances the interaction between the TCA cycle and the MAS, therefore, allows the metabolic status in the cytosol to be translated into the mitochondria and vice versa.
In conclusion, we propose that the elevation of cytosolic aspartate levels drive the MAS resulting in elevated ATP levels and 2-oxoglutarate dehydrogenase activity which causes increased levels of oxidative stress in kidney cells within 30 min of exposure. This elevation in dehydrogenase activity was shown in the MTT assay seen after 1h of exposure with aspartame. To further cement this, MAS activity measured in a whole cellular system needs to be assessed using radiolabeled glucose and measuring its uptake in the presence and absence of AOA (24).
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Maria A, Maryam A, Shoba VA (2019) Mitochondria-Derived Oxidative Stress Associated with Aspartame Effect on Kidney Cells. Int J Endocrinol Metab Disord 5(3): dx.doi.org/10.16966/2380-548X.161
Copyright: © 2019 Maria A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access