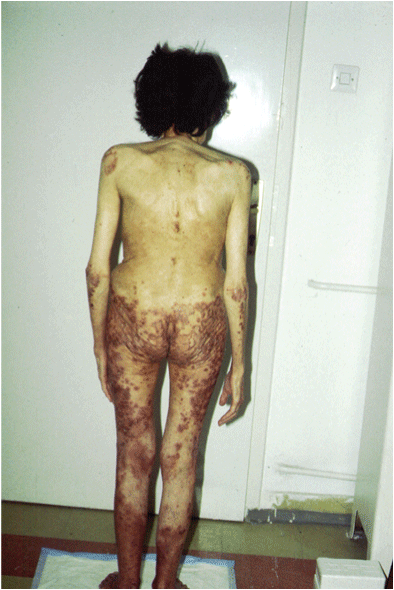
Figure 1: Erythematous papules with blistering, crusting and scaling at the borders on the buttocks and elbows.

Cedomir M Dimitrovski1* Nebojsa Pesic2 Jovan G Mircevski3 Gjorgi Gocev4
1Department of Endocrinology, Acibadem Sistina Hospital, Republic of Macedonia*Corresponding authors: Cedomir M. Dimitrovski, Professor, Department of Endocrinology, Acibadem Sistina Hospital, Skupi 5A, 1000 Skopje, University Clinical Center, Republic of Macedonia, E-mail: cedomir.dimitrovski@acibademsistina.mk
Objective: To describe a case of pancreatic glucagonoma with normal glucose tolerance and the difficulties of its early diagnosis and treatment.
Methods: We present the clinical, biochemical, imaging, and histopathological findings in a patient with glucagonoma syndrome.
Results: A 55-year-old cachectic woman with a long history of dermatosis was referred for evaluation of a pancreatic mass. The clinical manifestations of glucagonoma syndrome (necrolytic migratory erythema; cachexia; angular cheilitis; glossitis; normochromic-normocytic anaemia and hyperglucagonaemia) had been present at least 2 years before the correct diagnosis was established. An abdominal and endoscopic ultrasound showed a pancreatic hypoechogenic abnormality and a coronal T1 -weighted magnetic image of abdomen showed a low-intensity abnormality (3.0 cm by 2.5 cm) in the tip of the pancreatic tail that lied directly in the splenic hilum. The first pancreatic surgery was unsuccessful and she underwent a second one when the tumor was successfully removed. Three months after the operation the glucagon levels fell to normal and skin rash completely resolved. Healing resulted in hyperpigmentation. The histopathological findings (H&E and immunocytochemistry) confirmed a pancreatic islet α-cell tumor-glucagonoma. Eight years later, she is doing fine with no clinical, biochemical or radiological signs of glucagonoma recurrence.
Conclusion: The pancreatic glucagonoma cannot be easily diagnosed. Many of the symptoms are non-specific. However, the presence of necrolytic migratory erythema is characteristic of the glucagonoma syndrome and is the clue which leads to correct diagnosis. We emphasize the absence of diabetes or impaired glucose tolerance in our patient and surgical difficulties in finding a “hidden” glucagonoma at the tip of the pancreas tail that lied directly in the splenic hilum.
Glucagonoma; Erythema necrolyticum migrans; Glucose tolerance; Pancreatic surgery
Glucagonoma is one of the most rare neuroendocrine neoplasms of pancreatic islets. The cutaneous manifestations associated with tumors of the pancreatic islets were for the first time described in 1942 [1], but only in 1966 it was established that the syndrome is connected with hyperglucagonemia [2]. In 1974 Mallison et al. [3], on the basis of nine cases defined the syndrome of glucagonoma, where besides the tumor of α2 -cells of the pancreatic islets he also included: necrolytic migratory erythema, cachexia, glucose intolerance and anemia. It has been estimated that the glucagonomas occur at a frequency of 1 per 20 million persons per year [4].
The patient is a 55 year-old-female, a retired teacher, a widow and a mother of two children. Her disease began in 1999 and the first manifestations were cutaneous changes on the lower legs, the upper part of the feet and palms. The changes were skin blush with bubbles, rarely filled with blood content. The bubbles dried very fast and peeled together with the blushed skin in the course of 2-3 weeks. The skin changes were preceded by a strong itch. After a skin biopsy the disease was diagnosed as contact dermatitis manum et pedum. Adequately, she was treated with local corticosteroids and antihistaminics. However, in the course of the time the changes extended on the upper parts of the extremities, the chest, the abdomen and the genital region. All the time the angles of the mouth were chapped, the tongue was distinctly red, shiny and smooth.
Because of deterioration of the skin changes, the patient was again hospitalized at the clinic of dermatology and the second skin biopsy was performed. The histopathological features were hyperkeratosis, spongiosis, epidermal necrosis and edematous keratocytes. All these findings were compatible with Necrolytic Migratory Erythema (NME). The patient was suspected of having a pancreatic tumor and referred at the clinic of gastroenterlogy. The trans abdominal and endoscopic ultra sonography showed a tumorous formation in the pancreas tail with dimensions 3.0 by 2.5 by 2.5 cm. The fine needle biopsy of the pancreatic lesion was performed, but the cytologic finding was not in favour of pancreatic neoplasm. During 1999, the plasma glucagon levels were determined twice. The values were 386 ng/L and 290 ng/L (normal adult values 60-177 ng/L), respectfuly. Due to the justifiable suspicion of pancreatic glucagonoma, the patient underwent abdominal laparotomy. The operation was unsuccessful because during the exploration of the pancreas, the tumor was not found. The next two years she continued to lose weight and from 62 kg she went to 36 kg.
In 2001 the patient was referred to the endocrine clinic for evaluation of a suspected pancreatic glucagonoma. On physical examination, the patient was conscious, active, cachectic with supra umbilical cicatrix of about 30 cm long from the previous surgery. She was 150 cm tall, weighed 36 kg and her body mass index was 16 kg/m2 .
The dermatological status was characterized with general paleness and decreased skin turgor. Distally, on the lower and upper extremities, genito-femorally, perianally-gluteally and submammary, erythemtous changes of annular type were noticed. The lesions were 1-2 cm in diameter, individual, but frequently confluent, lightly erosive and serpiginous, with clearly demarked edges. On the fresh skin changes, a central vesicle was noticed filled with sero-hemorrhagical content. The skin changes were in different stages. At the same time, erythemo-desquamative, vesicular, necrotic and crusty changes were present on different places (Figure 1). The volar surface of the palms was diffusely erythemo-desquamative with scaling on the fingers. The nails were with no specific changes. The hair was dry and brittle.
Figure 1: Erythematous papules with blistering, crusting and scaling at the borders on the buttocks and elbows.
The histopathological features of cutaneous biopsy were compatible with necrolytic migratory erythema (Figure 2).
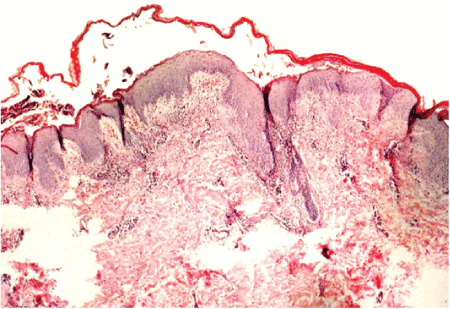
Figure 2: There is necrosis of the upper layers of the stratum spinosum and detachment from the subjacent viable epidermis. Also there is edema of the papillary dermis and infiltrates containing many neutrophils.
The results of pertinent investigations (with reference ranges shown parenthetically) were as follows: erythrocyte sedimentation rate 100 mm/h (12-19); C-reactive protein 10.5 mg/L (0-6); hemoglobin 10 g/dL (13.2-16.5); hematocrit 27% (38%-32%); leukocyte count 5.1 × 103 /µL (4- 11 × 103 ); platelet count 345 × 103 /µL (140-350 × 103 ); total cholesterol 2.8 mmol/L (up to 5.5 mmol/L); LDL-cholesterol l2.6 mmol/L (2.2-3.7); HDL-cholesterol 0.78 mmol/L (0.9-2.0); triglycerides 0.34 mmol/L (up to 2.0). Urea (serum) 6.0 mmol/L (2.7-7.8), creatinine 70 µmol/L (45-109); urinalysis norman; total serum protein 5.8 g/dL (6.4-8.3); albumin 2.8 g/ dL (3.4-4.7); globulin 2.5 g/dL (2.7-3.5). The fasting plasma blood glucose levels were normal on several occasions, 4.7 mmol/L, 5,1 mmol/L and 4,9 mmol/L, respectfully. A standard 2 hour oral glucose tolerance test (OGTT) with 1.75-g/kg of ideal body weight was performed and it was normal: glycemia (mmol/L) at 0 min 4.8; at 60 min. 5.1 and at 120 min. 4.5, respectfuly. The fasting serum insulin levels were 9.5 µU/ml and two hours after a meal 45.8 µU/ml (normal values 7 to 24). The baseline serum C-pepdide level was 1.7 ng/mL (1.0-4.0).
The plasma glucagon was measured using a comercial RIA (Linco Research Inc., St.Charles, Mo.). The basal plasma value was 540 ng /L (60- 177 ng/L).
Alanine aminotransferase, aspartate aminotransferase and lactate dehydrogenase were normal. The tumor markers: alfa fetoprotein, carcinoembryonic antigen and CA 19-9 were normal.
MRI of the upper abdomen in T1 and T2 pulse sequences, on the coronal T1-magnetic pictures a low-intensity abnormality (3.0 cm by 2.5 cm) was seen at the tail of the pancreas that lied directly in the splenic hilum. On T2 pictures after the applied contrast, the abnormality had a high-intensty (Figure 3).
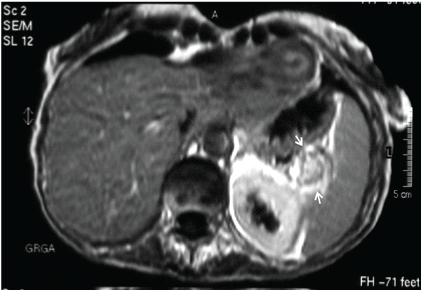
Figure 3: MRI of the upper abdomen: T2-pulse sequence after i.v. contrast. The pancreatic tumor is shown as a high-intensity round lesion (3.0 by 2.5 cm) in the region of the splenic hilum.
The diagnosis of the pancreatic tumor was confirmed and the patient underwent second laparotomy. This time distal pancreatectomy with splenectomy was performed. The pathohistological analysis of the surgical specimen confirmed the tumor cells of the pancreatic islets (Figure 4). The immune cytochemical staining confirmed the presence of glucagon in the tumor cells (Figure 5).
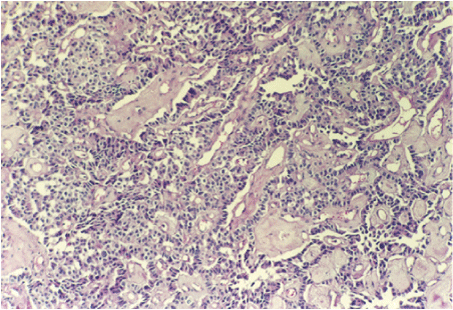
Figure 4: Histological appearance of the pancreatic tumor cells forming mixed trabecular and solid pattern with areas of hyaline stroma (H&E; original magnification × 40).
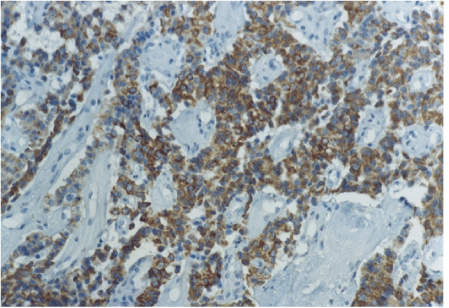
Figure 5: Immunohistochemical analysis of the pancreatic glucagonoma showing intense immunoreactivity for glucagon (Original magnification × 200).
Three months after the operation the patient felt quite well, got on weight 7 kg, all skin lesions disappeared, but with residual hyper pigmentation on certain places. The plasma glucagon level was in the normal range (86.0 ng/L).
The clinical analysis of our case shows that it is a classical case of glucagonoma syndrome that had been present at least two years before it was correctly diagnosed and surgically treated.
The pancreatic glucagonoma of our patient was accompanied by: necrolytic migratory erythema (NME), cachexia, cheilosis, glossitis, anemia, hypolipemia, hypoproteinemia, hyperglucagonemia, but normal glucose tolerance.
Besides these signs, the glucagonoma syndrome can be also accompanied by: gastrointestinal symptoms and signs (abdominal pain, anorexia, diarrhea and constipation); neuropsychiatric changes (ataxia, demention, optic atrophy, weakness of proximal muscles, psychosis and depression) and tendency to vein thrombosis [5,6]. The biochemical abnormalities can show hypo aminoacidemia and higher levels of gastrin, insulin, serotonin, vasoactive intestinal peptide, calcitonine, pancreatic peptide and adrenocorticotropic hormone. All these hormones can be secreted by the glucagonoma [7].
Besides the above-described classical syndrome, the pancreatic glucagonoma can be manifested in a second form only as a severe diabetes mellitus and in a third form together with multiple endocrine neoplasia type 1 [8]. Making the correct and timely diagnosis of glucagonoma is not an easy task, because most of the symptoms and signs are non-specific. Therefore, the diagnosis is made relatively late, usually one to six years after the appearance of the cutaneous manifestations [9].
The glucagonoma should continue to be searched if there is a constellation of the following symptoms and signs: loss of weight, NME, diabetes mellitus or impaired glucose tolerance. These changes are present in more than 70% of the patients [5]. The determination of glucagon in the blood is of special importance when NME and diabetes mellitus are present. The hyperglucagonemia less than 500 ng/L may be also found during being on a diet, in trauma, sepsis, hepatic and renal failure or other neuro-endocrine tumors, but values higher than 1000 ng/L are nearly diagnostic for glucagonoma [5].
Nonetheless, NME is the most characteristic sign of glucagonoma syndrome, but is not completely pathognomonic. NME or lesions similar to NME can be noticed in: zinc deficit, pellagra, the final stage of hepatic insufficiency, pemphygus foliaceus, toxic epidermal necrolisis and pustular psoriasis.
The hyperglucagonemia itself might be a cause for NME [10], as well as the hypoaminoacidemia. The parenteral nutrition with amino acids leads to the retreat of the skin lesions [11]. Due to the skin lesions, the first doctor who will most probably meet the patient and who should think of glucagonoma is the dermatologist. Exactly that was the case with our patient.
Most of the clinical signs of the glucagonoma syndrome (loss of weight, hypoproteinemia, hypolipemia and anemia), can be explained with the physiological catabolic effects of the hormone glucagon itself.
One of the characteristics of glucagonoma syndrome is also diabetes mellitus or carbohydrate intolerance.
Shah et al. [12] reported impaired glucose tolerance and lack of glucagon suppressibility after oral glucose load. On the other hand, Vinh et al. [13] demonstrated suppressability of glucagon in a patient with impaired glucose tolerance. Proglucagon may play role on the degree of glucose intolerance.
But, not all patients have diabetes or carbohydrate intolerance. Kindmark et al. [14] reported on series of 23 glucagonoma patients that only 22% of their patients had developed diabetes prior to the diagnosis of glucagonoma. Our patient was such a case.
During the eight years of follow-up the carbohydrate tolerance was preserved in our patient. It is well known that plasma concentrations of glucagon do not have best correlation with glucose tolerance (some patients with light hyperglucagonemia suffer diabetes, while the others with high hyperglucagonemia have a normal glucose tolerance). This knowledge is quite compatible with ours for the mutual relation of the glucagon and the insulin. Namely, on the liver level the glucagon causes glycolgenolisis (transitorily) and glyconeogenesis (persistently). From the other side, insulin inhibits the hepatic glucose production and stimulates the peripheral one (muscles, fatty tissue) glucose consumption. Whether the hyperglucagonemia will cause glucose intolerance depends on the magnitude of the hyperglucagonemia and the patient’s insulin reserves [15]. If the insulin reserves are intact, the increased release of insulin may compensate for the increased hepatic glucose production and the glucose tolerance will be increased. It is most probable that this mechanism operated in our patient and that is the reason she had a normal glucose tolerance.
The existence of clinical and/or laboratory symptoms and signs suspicious for glucagonoma need an application of imaging methods to localize the tumor. Most frequently applied are transabdominal and endoscopic ultrasonography, abdominal CT and MRI. These methods were also applied in our case, and the tumor was successfully localized by the transabdominal and endoscopic ultrasonography and MRI. Besides these methods, others that can be applied are: angiography, octreotide scintigraphy, but the best method for preoperative localization of the pancreatic glucagonoma is the endoscopic ultrasonography. Its sensitivity is 80%, and the specificity 95% [16].
From biological point of view, the big glucagonomas (>5.0 cm) are malignant in 60% to 80%, and the small ones (0.5 cm to 3.0 cm) are benignant [16]. But, histopathologically it is very difficult to define which is benignant and which one a malignant glucagonoma. Most frequent sites of glucagonoma metastasis are liver, regional lymph glands, bones, suprarenal glands, kidneys and lungs.
Surgery is a treatment of choice of glucagonoma. Depending on the place and the expansion of the tumor during the laparotomy, the surgeon may decide on a simple enucleation, focal pancreatic resection or Whipple’s procedure [17]. Our case was treated with distal pancreatectomy with splenectomy because of the location of the glucagonoma. It was at the tail of the pancreas that lied directly in the splenic hilum itself. Such “hidden” location of the glucagonoma is probably the cause why the surgeon did not succeed to find the glucagonoma during the first laparotomy.
In case of a metastatic disease and/or inoperability, chemotherapy is applied (streptozotocin; doxorbucin; fluorouracil; chlorozotocin; dacarbazin), hemoembolization and subcutaneous application of octreotide (somatostatin analogous). Due to the indolent growth of glucagonoma, even patients with metastatic disease survive longer [4,5].
We presented a 55 year-old-female with pancreatic glucagonoma who was successfully surgically treated. The clinical manifestations of glucagonoma syndrome (necrolytic migratory erythema; cachexia; angular cheilitis; glossitis; normochromic-normocytic anaemia and hyperglucagonaemia) had been present at least two years before the correct diagnosis was established. There was no impairment of glucose tolerance. An abdominal and endoscopic ultrasound showed a hypoechogenic abnormality and a coronal T1-weighted magnetic image of abdomen showed a low-intensity abnormality (3.0 by 2.5 cm) in the tail of the pancreas, lying at the splenic hilum. The second pancreatic surgery was successful and the tumor was removed. The histopathological findings (H&E and immunocytochemistry) confirmed a pancreatic islet α2 -cell tumor-glucagonoma.
Three months after the operation the glucagon levels fell to normal and skin rash completely resolved. Healing resulted in hyperpigmentation.
Eight years after the glucagonoma removal, there are no signs of metastasis in our patient.
Download Provisional PDF Here
Article Type: Case Report
Citation: Dimitrovski CM, Pesic N, Mircevski JG, Gocev G (2017) Pancreatic Glucagonoma with Normal Glucose Tolerance. Int J Endocrinol Metab Disord 3(3): doi http://dx.doi.org/10.16966/2380-548X.141
Copyright: © 2017 Dimitrovski CM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access