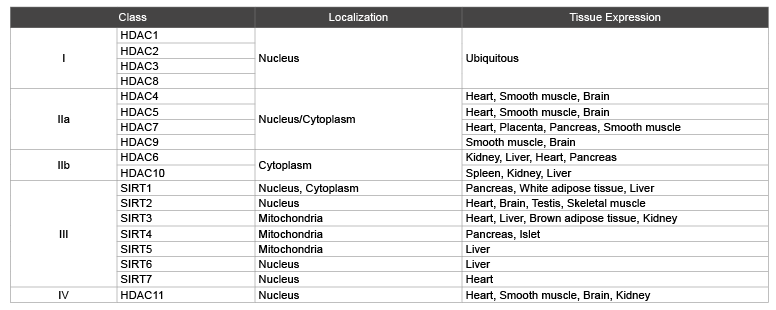
Table 1: Types of HDACs

Fenting Liu Zhemin Shen Heng Zhang Geyang Xu*
Department of physiology, School of Medicine, Jinan University, Guangzhou, Guangdong, China*Corresponding author: Geyang Xu, Department of Physiology, School of Medicine, Jinan University, 601 Huangpu Avenue West, Tianhe District, Guangzhou, Guangdong, 510632, China, Tel: 0086-20-85220260; E-mail: xugeyangliang@163.com
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) maintain the balance of histone acetylation, and play important role in chromosome modification and gene expression. Histone deacetylases have been identified to modify acetylation of proteins other than histones and inhibit gene expression. Here we summarize the functions of histone deacetylases in glycometabolism including the expression of histone deacetylases in different energy conditions, effects of histone deacetylases on islet development, glucose uptake in skeletal muscle, hepatic gluconeogenesis as well as the role of histone deacetylases inhibitors in glycometabolism. Modulation of histone deacetylases provides an alternative opportunity of a therapy for both type 1 and type 2 diabetes.
Histone deacetylases; Histone deacetylases inhibitors; Glucose metabolism; Type 1 diabetes; Type 2 diabetes
Epigenetics describes changes that are stable, but potentially reversible alterations in gene expression, that occur without permanent changes in DNA sequence and can still be passed on from generation to generation. Epigenetics portfolio includes products for studying DNA methylation, histone modification, genomic imprinting maternal effects, gene silencing and non-coding RNA etc. [1]. Histone modification is an especially important mode of epigenetic modifications. The amino-terminal tails of the histones are subject to post-translational modification by acetylation of lysine, methylation of lysine and arginine, phosphorylation of serine and ubiquitination of lysine [2]. Histone deacetylases (HDACs) and histone acetyl transferases (HATs) determine the pattern of histone acetylation, which together with other dynamic sequential post-translational modifications might represent a ‘code’ that can be recognized by nonhistone proteins forming complexes involved in the regulation of gene expression [2]. Under the effect of HAT, the acetyl of acetyl coenzyme A will be transferred to the ε-amidogen in N-terminal of lysine of histone protein to neutralize its positive charge and reduce the affinity between histone and DNA to activate gene transcription [3]. In contrast, HDACs make closely combine the deacetylation histone with the DNA phosphate backbone while the former contain positive charge and the latter contain negative charge. Making the chromatin structure dense can hinder combination of transcription factors and DNA so as to inhibit the gene transcription [4].
The activities of HATs and HDACs can be regulated by various signal transduction pathway. Recent studies found that abnormal regulation of epigenetic processes in the early life environment may lead to a susceptibility which increases the risk to later specific environmental exposures, associated with various metabolic disorders, such as cancer, cardiovascular disease, obesity and type 2 diabetes [1,5-7]. In this review we summarize the roles of HDACs in glucose homeostasis.
Histone deacetylases comprise a family of 18 genes, which are grouped into classes I–IV based on their homology to their respective yeast orthologues. Class I, II, and IV consist of 11 family members that are referred to as ‘classical’ HDACs, whereas the 7 class III members are called sirtuins (Table 1).
HDACs family mainly locate in the nucleus and cytoplasm, only a few of them in cytoplasmic organelles such as SIRT3 and SIRT4, SIRT5 of class III HDACs which position is in the mitochondria [2,8].
It is supposed that different energy state may change the gene expression profiles of HDACs. A study shows that the expression of hypothalamic HDACs is closely related to energy state. 16-hours fasting increased HDAC3 and HDAC4 expression levels in the medial hypothalamus compared to those of normally-fed mice, whereas HDAC10 and HDAC11 expression were decreased. Fasting did not alter the expression of HDAC1, −2, −5, −6, −7, −8 or, −9. Among all HDACs examined, a highfat diet significantly increased the expression of hypothalamic HDAC5 and HDAC8 [9]. Ma et al. [10] reported that alterations in HDACs in stomach under positive and negative energy states. Fasting stimulates HDAC4 but inhibits HDAC5. When animals were exposed to high fat diet, gastric HDAC4 was decreased, while HDAC5 was increased [10]. These observations suggest that HDACs are critically involved in energy metabolism.
Pancreatic islets are the regions of the pancreas that contain its endocrine cells, which play a significant role in diabetes. HDAC II (HDAC4, HDAC5 and HDAC9) regulates differentiation and development of the pancreatic cells [11]. Class IIa HDACs display a restricted expression pattern in the pancreas, with HDAC4, -5, and -9 specifically expressed in β cells. Overexpression of HDAC4 or HDAC5 in the pancreas caused a decline in β cell development, whereas mice lacking HDAC5 or HDAC9 in the pancreas have enhanced β cell mass, further supporting the role of class IIa HDACs in the control of the β cells lineage [12]. On the other hand, development of the endocrine cells in pancreatic islets can be promoted by HDAC II inhibitors. Treatment of pancreatic explants with the selective class IIa HDAC inhibitor MC1568, which modulates the stability of class IIa HDAC-MEF2 complexes, enhances expression of Pax4, a key factor required for proper β-and δ-cell differentiation and amplifies endocrine β [13,14].
Table 1: Types of HDACs
HDAC I: Inhibition of HDAC I plays an essential role in the carbohydrate metabolism in skeletal muscle. Synthetic class I HDAC inhibitors can enhance mitochondrial function. Treatment with global or class I HDAC inhibition resulted in increased mitochondrial density and activity in C2C12 myotubes, while inhibition of class II HDACs had no effect on these parameters. Global or class I selective HDAC inhibition increased expression of several key mitochondria-related transcription factors, as well as the levels of multiple genes involved in glucose and lipid metabolism [15,16].
HDAC II: Skeletal muscle is a major site of glucose uptake after insulin administration. Insulin stimulation leads to an uptake of glucose through Akt mediated translocation of glucose transporter 4 (GLUT4) to the cell surface [17]. Although skeletal muscle (GLUT4) expression is not compromised in diabetes and obesity, insulin resistance associated with obesity and diabetes is ameliorated by specific overexpression of GLUT4 in skeletal muscle. Interestingly, this effection can be reversed by HDAC5 [18]. It is reported that AMP-activated protein kinase (AMPK) regulates GLUT4 transcription through the histone deacetylase (HDAC5) transcriptional repressor [18]. Knockdown of HDAC5 in human primary muscle cells increased glucose uptake and was associated with increased GLUT4 expression but reduced GLUT1 expression [19]. Inhibition of HDAC5 enhances metabolism and insulin action in muscle cells, as these processes in muscle are dysregulated in metabolic disease, HDAC inhibition could be an effective therapeutic strategy to improve muscle metabolism in metabolic diseases such as obesity and diabetes [18,19].
HDAC III: SIRT6, a member from HDAC III, maintains efficient glucose flux into tricarboxylic acid cycle (TCA cycle) under normal nutrient conditions. Zhong et al. [20] showed that SIRT6 deficient mice exhibited a pronounced increase in glucose uptake both in muscle and brown adipose tissue, whereas no changes were observed in the liver, brain or heart [20]. SIRT6 influence glycolysis as a co-repressor of Hif1α, which acts as a safeguard mechanism to down-modulate basal transcription of Hif1α target genes under normal nutrient conditions [21].
In HepG2 cells and mouse livers, the expressions of phosphoenol pyruvate carboxykinase (PEPCK) and hepatocyte nuclear factor 4a (HNF4a) were significantly decreased by treatment with a newly designed class I HDAC inhibitor, Ky-2 [22]. SiRNA knockdown of HDAC1 expression, but not of HDAC2 or HDAC3, in HepG2 cells also restrained PEPCK and HNF4a expression [23]. In addition, HDAC6 is an essential regulator of hepatic glucocorticoid-stimulated gluconeogenesis and impairment of whole-body glucose metabolism through modification of glucocorticoid receptor nuclear translocation. Selective pharmacological inhibition of HDAC6 may provide a future therapeutic option against the prodiabetogenic actions of glucocorticoids [24].
Ghrelin and NUCB2/nesfatin-1: Gastric hormones ghrelin and NUCB2/nesfatin-1 are secreted from X/Alike cells in the fundus of stomach. Previous studies suggested that expression of gastric ghrelin and NUCB2/nesfatin-1 are reciprocally related to organism energy supply [25]. Ma et al. reported that HDAC5-mTORC1 signaling as a novel mechanism in the differential regulation of gastric ghrelin and nesfatin-1. Sodium valporate (VPA), a broad-spectrum inhibitor of histone deacetylases (HDACs), increased ghrelin while decreased nesfatin-1 in mice fed normal chow (NCD) or high fat diet (HFD). Alterations in ghrelin and NUCB2/nesfatin-1 were mediated by HDAC5 but not HDAC4 [10].
FGF21: Fibroblast growth factor 21 (FGF21) was originally identified as a member of the FGF family in homology studies and is a member of the endocrine FGF subfamily that lacks heparin binding domains and is released into the circulation. A potential role as a metabolic regulator emerged when FGF21 was shown to increase glucose uptake in adipocytes [26]. Moreover, Previous studies showed that serum levels in FGF21 are increased in T2D patients and correlated with fasting insulin and BMI, which also has a direct effect in enhancing skeletal muscle glucose uptake and improved the lipid profile [27,28]. Therefore, FGF21 is defined as a novel metabolic regulator shown to improve glycemic control [29]. Short free fatty acid butyrate stimulates FGF21 expression through activation of PPAR-α. The PPAR-α activation is a result of HDAC3 inhibition by butyrate, thus inhibition of HDAC3 represents a new approach in the induction of FGF21 [30]. Moreover, Li Y et al. found that SIRT1 is a major regulator of fasting-inducible FGF21. Hepatic SIRT1 and FGF21 both are physiologically regulated by a feeding-fasting-refeeding cycle, SIRT1 deficiency interfere with fasting-induced production and release of hepatic FGF21 [31].
In summary, studies indicate that HDACs differentially regulates the synthesis and release of metabolic hormones, which subsequently plays an important role in the maintenance of organism energy homeostasis. Thus modulation of specific HDAC isoforms may thus be a potential target for treatment of obesity and its associated metabolic diseases such as diabetes.
The term ‘HDAC inhibitors’ is commonly used for compounds that target the ‘classical’ class I, II, and IV HDACs and that are currently evaluated in clinical trials [32]. Recent evidences suggested that there is a link between diabetes and HDACs, because HDAC inhibitors promote beta-cell development, proliferation and function as well as improve glucose homeostasis. Treatment of obese diabetic mice with a class I– but not a class II–selective HDAC inhibitor enhanced oxidative metabolism in skeletal muscle and adipose tissue and promoted energy expenditure, thus reducing body weight and glucose and insulin levels. These effects can be ascribed to increased PGC-1α action in skeletal muscle and enhanced PPARγ/PGC-1α signaling in adipose tissue. These results suggest that class I HDAC inhibitors may provide a pharmacologic approach to treating type 2 diabetes [15].
Trichostatin A: TSA significantly enhanced insulin stimulated glucose uptake, glycogen synthesis and glycogen synthase activities in C2C12 myotubes. Insulin stimulated phosphorylations in insulin receptor; Akt and GSK3β were remarkably increased in the TSA-treated cells. These improving effects of TSA were probably due to HDAC2 inhibition, since the enhanced expression of HDAC2 could abolish the TSA-induced improvement in the insulin signaling transduction. These data suggest that HDAC2 should be an important potential target for regulating insulin sensitivity [33,34].
Sodium butyrate (NaB): Sodium butyrate (NaB) is a short chain fatty acid having HDAC inhibition activity. NaB treatment decreased plasma glucose, HbA1c, beta-cell apoptosis and improved plasma insulin level and glucose homeostasis through HDAC inhibition and histone acetylation in diabetic animal as compared to control. NaB treatment improved the beta-cell proliferation, function and glucose homeostasis as well as reduced beta-cell apoptosis in juvenile diabetic rat by the modulation of p38/ERK-MAPK and apoptotic pathway [35].
Synthetic HDACs inhibitor: A significant reduction of body weight, fasting glycemia in animals treated with SAHA (pan-inhibitor) or MS275 (a class I HDAC selective inhibitor) but not in those treated with MC1568 (a class II HDAC inhibitor). Moreover, global or class I–selective HDAC inhibition improved glucose clearance. Circulating triglycerides and nonesterified fatty acids were also decreased after SAHA and MS275 treatment. Moreover, MS275 completely cleared the lipids that accumulate in the liver of db/db mice, while SAHA had a significant but milder effect [15].
HDACs are involved in several biological pathways relevant for the etiology and pathogenesis of type 1 and type 2 diabetes [36]. Modulation of histone deacetylase (HDAC) activity has been implicated as a potential therapeutic strategy for type 1 and type 2 diabetes.
The level of HDAC2 and HDAC3 in islets of patients with type 1 diabetes is remarkably higher than in islets in non-diabetic individuals, while HDAC1 is less in diabetic patients than in islets of non-diabetic persons, which confirms that HDACs indeed have regulatory effects on these islets [37]. Further exploration suggests that specific inhibition of HDAC3 protects β-cells from cytokine-induced apoptosis, an important event in the development of type 1 diabetes [38]. In animal models, mice did not show hyperglycemia when subject to coinjection of streptozotocin and HDACs inhibitor ITF2357 [39]. On the other hand, the pathogenesis of type 2 diabetes involves a combination of peripheral insulin resistance and pancreatic β-cell failure. Data from the literature indicate that HDAC3 regulates genes involved in key metabolic events. HDAC3 inhibition improves hyperglycemia, insulin secretion and pancreatic insulin content in a rat model of type 2 diabetes and in db/db mice, indicating selective inhibition of HDAC3 may be an attractive strategy for targeting type 2 diabetes [16].
Changes in human behavior and lifestyle over the last century have resulted in a dramatic increase in the incidence of diabetes worldwide. The epidemic is chiefly of type 2 diabetes and also the associated conditions known as ‘diabesity’ and metabolic syndrome [40]. About one-third of a billion of world population is now living with diabetes and it continues to increase in numbers and significance [41]. Novel evidence suggests that HDACs are critical regulatory molecules in glucose metabolism. Further studies are needed to provide new information on the integration of cellular activities of HDAC isoforms with overall glucose metabolism. Results of these new investigations may yield new insights relevant to treatment strategies for human diabetes.
Supported by grants from the National Natural Science Foundation of China (31401001), the Science and Technology Planning Project of Guangdong Province, China (2014A020212210), the Guangdong Medical Science Research Foundation (A2014375), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (20141685) and special Grants from the Guangzhou Pearl River Young Talents of Science and Technology (201504281553023).
Download Provisional PDF Here
Article Type: Review Article
Citation: Liu F, Shen Z, Zhang H, Xu G (2015) The Role of Histone Deacetylases in Glucose Metabolism: a Mini- Review. Int J Endocrinol Metab Disord 1(3): http://dx.doi.org/10.16966/2380-548X.113
Copyright: © 2015 Liu F, et al. This is an openaccess article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access