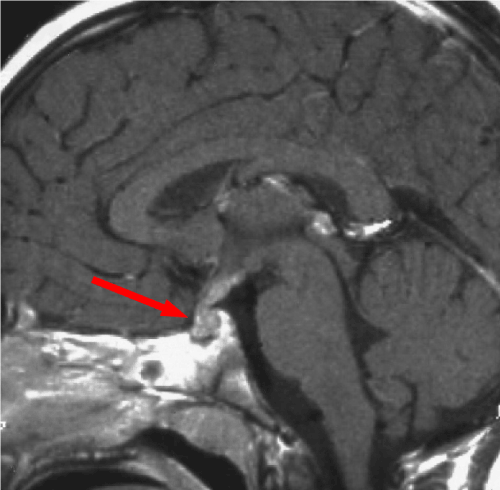
Figure 1: Coronal View of RM T1 post-gadolinium with enhancement of pituitary gland and its stalk

Chiloiro S* Giampietro A De Marinis L
Pituitary Unit, Departments of Endocrinology, Catholic University School of Medicine, Largo Agostino Gemelli, 8, 00168, Rome, Italy*Corresponding author: Sabrina Chiloiro, Pituitary Unit, Departments of Endocrinology, Catholic University School of Medicine, Largo Agostino Gemelli, 8, 00168, Rome, Italy, E-mail: schiloiro@gmail.com
Intracranial teratomas are rare; suprasellar teratomas can result in visual disturbance and in diabetes insipidus and hypopituitarism, and in case of suprasellar teratomas extended into the pituitary fossa. We report a case of 29-year female pan-hypopituitaric patient with suprasellar mature teratoma history, recently admitted to our Pituitary Disease Unit.
At admission in our department patient appeared vigilant, cooperative but showed impaired mental concentration and moderate disorientation. Patient’s weight was 100.5 kilograms, while her height was 170 centimeter (BMI: 34.8 Kg/m2). Neurological examination excluded visual abnormalities. Laboratory tests showed normocytic normochromic anemia and confirmed pan-hypopituitarism. Serum alpha-feto protein (AFP) and serum beta-human chorionic gonadotropin (beta-HCG) were normal. Bone densitometry (DEXA) documented lombar-spine osteopenia. Cranial and hypothalamic-pituitary contrasted MRI (c-MRI) was negative for teratoma recurrence. Abdominal ultrasound examination documented hepatic steatosis, electroencephalogram showed frontal and temporal abnormal slow waves and epileptiform abnormalities. Neuropsychological tests demonstrated alterations in memory and executive functions. Esophagogastroduodenoscopy documented antral gastritis, while colonscopy excluded stenosis and inflammation. Chest X-ray, echocardiography and thyroid ultrasound examinations were normal. Replacement hormone therapy (levothyroxine, hydrocortisone, transdermic norelgestromin, ethinylestradiol, desmopressin and colecalciferol) and medical treatment with carbamazepine and metformin were ongoing.
Clinical history started on 1991 when patient was 6-years old age and she was admitted to other hospital for polyuria, polydipsia, weight gain and linear growth reduction. Hormonal test revealed diabetes insipidus and hypopituitarism. Contrasted-MRI evidenced thickening of the pituitary stalk, low intensity suprasellar lesion, heterogeneous gadolinium-enhancement (Figures 1 and 2). Right sub-frontal biopsy was performed and histological examination was suggestive of germinoma tumor. On March 1991, conventional radiotherapy treatment (32.4 Grey to the whole brain and additional 21.6 Grey to the tumor) was performed. After 6 months for disease-persistence, on September 1991, stereotactic biopsy was performed and histological examination was suggestive of third ventricle choroid plexus papilloma. Patient underwent neurosurgical resection. Histological examination of the specimens documented cartilaginous tissue with nerve and muscle fibers, mucous tubules filled with mucus and epidermoid cysts, suggestive for mature teratoma.
Every 6 months, endocrinological evaluation and cranial-pituitary c-MRI were scheduled. At 10-years old age (on 1994), physical examination revealed a weight of 38 kg (90th percentile, Tanner-Whitehouse growth charts) and a height of 124 cm (3rd percentile). Velocity growth was about 3 cm/year (<3rd percentile). Patient target height was 171.5 cm (90th percentile). Basal hormonal evaluation confirmed hypopituitarism and c-MRI did not documented teratoma recurrence. Therefore, patient was tested with clonidine test for impaired GH secretion and diagnosis was confirmed. After three disease-free survival years (DFSY), on November 1994, rhGH replacement therapy (rhGH-RT) was started (somatropin 1.21 mg/daily). rhGH-RT was well tolerated and no adverse events occurred.
Figure 1: Coronal View of RM T1 post-gadolinium with enhancement of pituitary gland and its stalk
On 2003, cranial c-MRI evidenced a suspected falx cerebri meningioma and rhGH-RT was discontinued (patient age 18 years). Patient underwent radical neurosurgery. Pathological diagnosis was atypical meningioma. 1-year post-surgery c-MRI confirmed tumor radical resection. rhGH treatment was re-started. Every 6 months, endocrinological evaluation and cranial c-MRI were scheduled. Cranial c-MRI performed on April 2011 showed meningioma recurrence. Patient underwent second radical neurosurgery. Histological examination documented transitional meningioma with GH receptor, progesterone receptor, and estrogen receptor and showed immunohistochemical strong positivity, weak positivity and negativity, respectively. According to this findings, treatment with rhGH was discontinued (Patient age was 27 years). C-MRI scheduled every 6 months did not show recurrence disease.
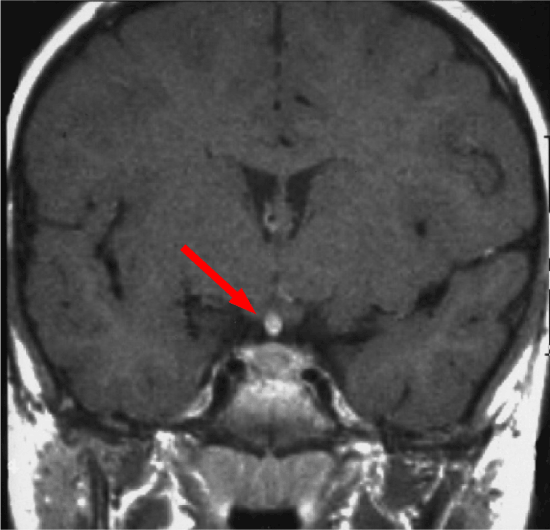
Figure 2: Sagital View of RM T1 post-gabolinium with enhancement of pituitary gland and its stalk
To our knowledge, our case is the first describing rhGH and estrogenprogestin replacement treatments in patient with teratoma history.
No evidence is actually available to rhGH-RT in teratoma. In our case, patient started rhGH-RT in other endocrinological specialistic center for severe growth delay and GH deficiency. In our teratoma case, rhGH-RT seems safe: during 10 years of rhGH-RT, teratoma recurrence did not occur. However, rhGH-RT was discontinued for recurrence of meningiomas with a strong GH receptor expression.
In vitro and in vivo xenobiotic studies demonstrated that GH and IGF-I increased DNA synthesis and consequently growth rate of meningiomas and that interruption of IGF-I receptor binding decreases meningioma growth In vitro [1].
Actually, association between rhGH-RT and malignancies is still controversial: in GH-treated Childhood Cancer Survivor Study (CCSS) participants, the relative risk of subsequent neoplasm (including CNS), was estimated at 2.15 with a long follow-up period [1]. In GH post marketing studies, overall 5-year cumulative incidence of subsequent neoplasms in CCSS estimated to be 6.2 [1]. Other authors have reported no association between GH replacement therapy and subsequent malignancies; Patterson et al. [1] showed a super imposable rate ratio for the development of any CNS subsequent neoplasm between childhood cancer survivors treated with GH as compared with those not treated. Mo et al. [2] observed a similar cancer incidence in 1204 children-onset GHD (CO-GHD) patients who received GH treatment as children as compared to general population. Moreover, also, cranial irradiation is a predisposing factor for subsequent intracranial tumors [3].
In a series of 60 adult rhGH replacement therapy patients treated with radiotherapy for primary brain tumors, meningiomas were diagnosed in three cases before starting rhGH treatment and in five cases during rhGH therapy. In this series, the finding of new second tumor is consistent with the well-established increased risk in all cancer patients of developing second CNS neoplasms. In particular, meningiomas usually occurred two or three decades after radiotherapy [4]. This finding is also confirmed by other authors: Mackenzie et al. [5] in a series, 220 GHD patients underwent cerebral irradiation for CNS primary tumors demonstrated that primary CNS recurrence risk and second neoplasm risk (as meningiomas and oligodendrogliomas) were similar in patient in rhGH replacement therapy (110 cases) as compared to patient who did not performed rhGH replacement therapy
Moreover, in a series of 416 CO-GHD patients 32 deaths were described. However, 93% of death cases occurred for CNS related cancer; of these 78% had undergone CNS irradiation, 22% had a primary diagnosis of malignant cancer and only 41% had received GH replacement treatment. Therefore, the authors suggested that, in this series, cancer death among CO-GHD patients were more likely due to primary disease and CNS irradiation and not to GH replacement treatment received [6].
In particular, meningiomas usually occurred two or three decades after radiotherapy [3]. Younger age at diagnosis, female gender, higher cranial radiation dose and a longer elapsed time since cranial radiation were associated with increased rates of meningioma, regardless of GH exposure [1]. In a series of 60 adult rhGH-RT patients treated with radiotherapy for primary brain tumors, meningiomas were diagnosed in three cases before starting rhGH-RT and in five cases during rhGH-RT [3]. The finding of new second tumor is consistent with the well-established increased risk in all cancer patients of developing second central nervous system (CNS) neoplasms [3].
However, as for safety reason, it is recommended that IGF-I levels should be maintained within age- and gender-based reference ranges, in pediatric patients treated with rhGH-RT: decreasing the risk of exposure to high IGF-I levels reduces the theoretical risk of cancer and other adverse events related to high IGF-I levels [7].
There are several inconsistencies in the literature about mitogenic effect of sex steroid hormones on meningiomas both in vitro and in vivo models [8]. Human meningioma tissues are mostly estrogen-receptor (ER) negative and progesterone-receptor (PgR) positive in ligand binding and enzyme immuno-assays.
Hsu et al. [9] detected PgR and ER in 83% and 8.6%, respectively, of 70 meningiomas. Hilbig et al. [10] reported that all the 116 cases of meningiomas were ER negative, whereas PgR was found in 58.3% of typical and in 48.2% of atypical meningiomas. The PgR expression and regulation in meningiomas is still unknown, since ERs are virtually absent. Several splice variants of ER mRNA have been identified in human meningioma tissues, including variants lacking exons 4, 5, and 7, but they are not involved in PgR expression.
In a series of 50 completely resected benign WHO I meningiomas, with and without recurrence respectively, progesterone receptor negativity had a strong predictive value of recurrence whereas the estrogen receptor status was not relevant to aid an accurate prognosis [8]. The lack of PgR is often associated with high tumor grade, increased mitotic activity and brain invasion, and represents an unfavorable prognostic factor for these tumors [8]. No conclusive data are currently available about the risk of meningioma in HRT users [8]. However, the high proportion of meningiomas in females, their accelerated growth during the luteal phase of the menstrual cycle and during pregnancy, and the association between meningioma and breast cancer have suggested that sex steroid hormones may be involved in the growth of these tumors [8]. Moreover, Patterson et al. [1] documented no association between estrogen and/or progesterone treatment and the development of meningioma as second malignancies in pediatric cancer survivors [1].
In our case, meningioma progesterone receptor immunohistochemical is weak positive and estrogen receptor immunohistochemical is negative. According to this finding, meningioma recurrences in our case seem not to correlate to puberty or HRT, because occurred when the patient was 27 years-old. Moreover, actually HRT is still ongoing, without any neuroradiological signs of meningioma recurrence.
In conclusion, in this paper, we report a case of a patient with mature suprasellar teratoma underwent first-line treatment with radiotherapy. This therapeutic change was justified by the suspected diagnosis of germinoma, which was excluded only through histological examination of the neurosurgery sample. In fact, routinely treatment for mature teratoma is neuro-surgical resection, because of their benign behavior. Radical excision is advocated, as long-term outcome is excellent [11]. Recurrence rate for mature teratoma is extremely low in cases of complete resection and usually occurs within 1 year after treatment [12].
Instead, radiotherapy is not appropriate in the therapeutic management of mature teratomas, which are considerate radio-resistant tumor, as for a low growth rate. This consideration is important in differential diagnosis and empirical treatment of sellar-suprasellar tumors, in order to minimize the risk of a secondary malignancy, particularly, in children that may need rhGH substitutive therapy.
In fact in the case we reported, early development and subsequent reoccurrence of meningioma occurred. In this case, meningioma seems, possibly, either primary or secondary to radiotherapy rather than to rhGH treatment, also according the patient young age, gender and the high radiation dose. A typical histology of the meningioma may also explain an accelerated growth rate as compared to less aggressive meningiomas that appear two to three decades after radiotherapy.
During post-treatment teratoma follow-up, in cases of healing, according to clinical and neuroradiological examination, hormone replacement therapy should be considered, also for GH deficiency. In fact, treatment of severe growth delay and GH deficiency with rhGH may benefit children treated for a mature intracranial teratoma. However, IGF- 1 levels should be intensively followed and should be maintained within age- and gender-based reference ranges, especially in irradiated children.
However, as actually few evidence are available, safety data have to be confirm also trough a multicentric prospective surveillance study.
The authors have not any conflict of interest and paper did not receive any financial grant.
Download Provisional PDF Here
Article Type: Case Report
Citation: Chiloiro S, Giampietro A, De Marinis L (2015) Safety of rhGH Replacement Therapy in Suprasellar Pituitary Teratoma: A Case-Report. Int J Endocr Metab Disord 1(2): doi http://dx.doi.org/10.16966/2380-548X.107
Copyright:© 2015 Chiloiro S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access