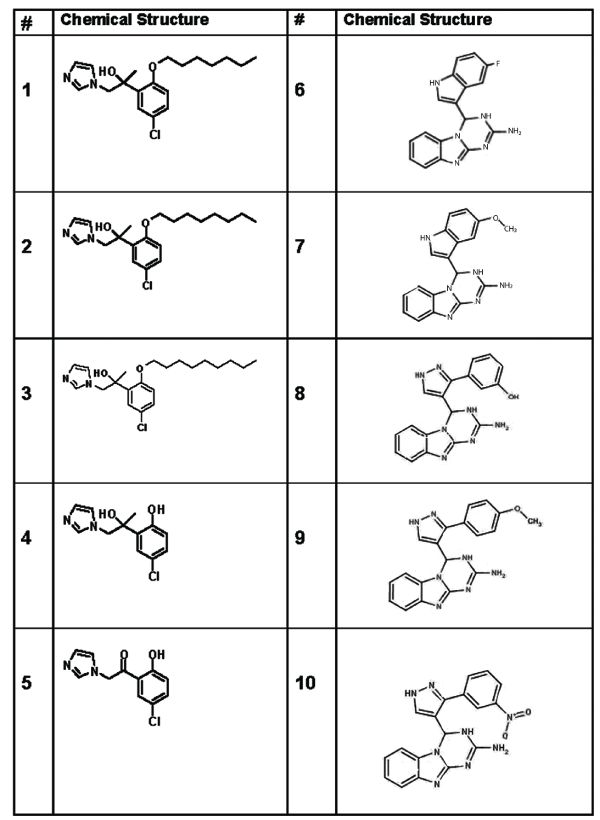
Figure 1: Chemical structures of ten small molecules with potential to interact with the G-CSF receptor.

Vasyl Sava1 Shijie Song1,2 X Kong3 Ghattas El Bassit1 Niketa Patel2,4 Kenyon G Daniel5 Said M. Sebti6 Juan Sanchez-Ramos1*
1Department of Neurology, University of South Florida, USA*Corresponding author: Juan Sanchez-Ramos, PhD, MD, Department of Neurology, University of South Florida, 13220 Laurel Drive, Tampa, FL 33612, USA, Fax: 813-974-8032; E-mail: jsramos@health.usf.edu
Small molecule mimetics of G-CSF may serve as potential drugs for neurodegenerative diseases, stroke and brain trauma.
Objectives: a) to identify drugs with potential to interact with the G-CSF receptor; b) to measure the binding parameters of these drugs with the G-CSF receptor expressed by neuronal and monocytic cell lines; c) determine whether these drugs trigger or block intracellular signaling elicited by G-CSF.
Methods: a) Computer-assisted modeling of 3 previously studied G-CSF mimetic drugs guided the search of chemical libraries for additional drugs with G-CSF receptor binding potential; b) G-CSF receptor binding parameters were assessed by measuring the capacity of the drugs to displace [125I]-G-CSF from its receptor in both monocytic and neuronal cell lines. c) The cellular response was determined by measurement of the expression of survival proteins Bcl2 and PKCδVIII protein with Western blot.
Results: The two most effective competitors for the G-CSF receptor triggered dose-dependent increases in Bcl2 expression in both the neuronal and monocytic cell lines. In the monocytic cell lines, these same two drugs were effective in blocking Bcl2 expression elicited by G-CSF.
Conclusions: Drugs that mimic the neurotrophic and/or immune-modulating actions of G-CSF will be useful as research tools, but may eventually be applied to clinical practice.
G-CSF receptor; Agonist; Antagonist; Monocytic cell line; Neuronal cell line; Signal transduction; Bcl2; PKCδVIII
Granulocyte colony-stimulating factor (G-CSF) is a hematopoietic cytokine commonly used for treatment of neutropenia and to increase generation of hematopoietic stem/progenitor cells in bone marrow donors [1,2]. G-CSF also exerts direct effects on neural stem/progenitor cells and is expressed, along with its receptor G-CSF-R, in neurogenic zones of the hippocampus (HC), the sub-ventricular zone and the olfactory bulb [3]. The G-CSF receptor and its ligand are also expressed by mature neurons in several other areas of the brain including pyramidal cells in cortical layers (specifically II and V), entorhinal cortex, Purkinje cells of the cerebellum, and in cerebellar nuclei in rats [3]. The G-CSF receptor can also be found on the cell surfaces of endothelial cells, lymphocytes, platelets, and neutrophils [4-6].
The role of G-CSF as a neurotrophic factor during development and in adult life is increasingly recognized. Mice bred to have a deficiency in G-CSF were reported to have problems with memory formation and development of motor skills [7]. More specifically, the hippocampus from these mice exhibited deficits in the induction of long term potentiation in the CA1 region, decreased neuronal precursor cells in the dentate gyrus (DG) and decreased dendritic complexity in neurons in the DG and CA1 region of the HC [8]. The defects seen in G-CSF deficient mice support the designation of G-CSF as a true neurotrophic factor, playing a role in neurogenesis and the maintenance of structural and functional integrity of the hippocampal formation. In combination with cognitive training, G-CSF can also significantly improve spatial learning and new neuron survival in the hippocampus [8].
The recognition that G-CSF acts directly on neural tissue has stimulated research on the therapeutic applications of these agents for neurodegenerative diseases, stroke and brain trauma [9-12]. G-CSF administration was reported to decrease amyloid burden, enhance neurogenesis, synaptogenesis and cognitive performance in a mouse model of Alzheimer’s disease (AD) [13]. Systemic G-CSF administration to rats that had sustained traumatic brain injury (TBI) resulted in significantly better motor function recovery than the control group [14]. More recent studies have reported that G-CSF administration to mice in a model of traumatic brain injury resulted in enhanced recovery of cognitive function in a hippocampal-dependent learning task [15-17]. G-CSF has also been reported to improve survival in a transgenic mouse model of amyotrophic lateral sclerosis- ALS [18]. In a mouse model of AD (the transgenic APP/PS1 mouse), G-CSF treatment was effective in decreasing amyloid burden, enhancing neurogenesis and improving performance on a hippocampal-dependent task [13].
There are several reasons for developing “druggable” G-CSF agonists and/or antagonists. G-CSF is a protein produced by recombinant DNA technology and hence is very expensive to manufacture. Therapeutic administration of G-CSF can produce serious adverse effects (thrombocytopenia, leukocytosis, anemia, and splenomegaly) from its peripheral actions on the bone marrow. Therefore, a drug which blocks the peripheral actions of G-CSF without blocking its central neurotrophic actions would be highly desirable for those at risk of vascular occlusions or hemorrhage. Also, a G-CSF mimetic drug that selectively stimulates brain G-CSF receptors might serve as a very useful agent to promote hippocampal neurogenesis and enhance cognition in Alzheimer’s disease.
Three small non-peptidergic molecules have been previously reported to act-like G-CSF; these drugs were shown to stimulate blood stem cell proliferation and to increase levels of circulating leukocytes in a rodent model [19]. However, the Kusano laboratory did not pursue this lead further. Other researchers have cloned, purified and crystallized the G-CSF-R to generate X-ray diffraction patterns that allow reconstruction and visualization in 3D computer models [20,21].
Coordinates for G-CSF bound to its receptor show a key point of interaction whereby a glutamate (E20) from G-CSF protrudes into a small pocket of GCSF-R, which is estimated to be a hydrogen bond accepting region (https://www.ncbi.nlm.nih.gov/protein/?term=1PGR). We hypothesized that a molecule capable of interrupting this interaction could reduce binding of G-CSF with G-CSFR.
The primary objectives of this study were a) to identify compounds with similar G-CSF receptor binding as the Kusano compounds; b) to measure the binding parameters of these small molecules with the G-CSF receptor expressed by neuronal and monocytic cell lines; c) determine whether these small molecules could trigger or block the anti-apoptotic intracellular signaling cascade elicited by G-CSF.
Two human cell lines were chosen for these cell culture studies. One is a human monocyte cell line that is representative of the peripheral immune system, THP-1, which grows in suspension. The other cell line is a human neuroblastoma cell line (SH-SY5Y) which is a mixed cell line although only adherent cells were utilized [22]. Both cell lines are known to express the G- CSFR [23,24]. Both cell lines were purchased from ATCC (Manassas, VA). THP-1 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) plus 1% penicillin-streptomycin (ATCC, Manassas, VA) in a humidified atmosphere of 5% CO2 at 37°C. SH-SY5Y cell line were cultured in 1:1 EMEM/F12 medium with 10% FBS plus 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
Compounds 1–3 (Table 1 and Figure 1) were originally discovered by Kusano and found to stimulate blood stem cell proliferation and to increase levels of circulating leukocytes in a rodent model [19]. The other drugs were identified with a limited computational analysis. Schödinger 3D-modeling software (https://www.schrodinger.com/smdd/) was used to import a molecular model of GCSF bound to its receptor from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/?term=1PGR). After modeling the first 3 molecules previously reported as potential G-CSF mimetics [19], eight other molecules were identified for study as potential inhibitors of the protein/protein interaction between G-CSF at its receptor site in cell cultures.
Figure 1: Chemical structures of ten small molecules with potential to interact with the G-CSF receptor.
| 1 | 2-(5-chloro-2-(heptyloxy) phenyl)-1-(1H- imidazol- 1-yl)propan-2-ol PubChem CID:10020811 | 6 | 4-(5-methoxy-1H-indol-3-yl)-3,4- dihydro- [1,3,5]-triazino-[1,2a] benzimidazol-2-amine PubChem CID: 5295059 |
| 2 | 2-(5-chloro-2-(octyloxy) phenyl)-1-(1H- imidazol- 1-yl)propan-2-ol PubChem CID: 10451448 | 7 | 3-[4-(2-amino-3,4-dihydro- [1,3,5] triazino[1,2- a]benzimidazol-4-yl)- 1H-pyrazol-3-yl]phenol PubChem CID: 5295059 |
| 3 | 2-(5-chloro-2-(nonyloxy) phenyl)-1-(1H- imidazol- 1-yl)propan-2-ol PubChem CID: 9907813 | 8 | 4-[3-(4-methoxyphenyl)-1H-pyrazol- 4-yl]-3,4- dihydro[1,3,5]triazino[1,2- a]benzimidazol-2- amine PubChem CID: 5295058 |
| 4 | 1-(5-chloro-2- hydroxyphenyl)-2-(1H- imidazol-1-yl)ethan-1-ol PubChem CID: NA | 9 | 4-(5-fluoro-1H-indol-3-yl)- 3,4dihydro- [1,3,5]triazino[1,2- a] benzimidazol-2-amine PubChem CID: 5295057 |
| 5 | 1-(5-chloro-2- hydroxyphenyl)-2-(1H- imidazol-1-yl)ethan-1-one PubChem CID: 12953655 | 10 | 4-[3-(3-nitrophenyl)-1H-pyrazol-4- yl]-3,4-dihydro[1,3,5]triazino[1,2-a]- benzimidazol- 2-amine PubChem CID: 5294981 |
Table 1: Chemical Names of Compounds Tested
Ten compounds were utilized for the present study (Table 1 and Figure 1). Compounds 1-5 were synthesized at the Drug Discovery Center (Moffitt Cancer Center) and Cmpds 6-10 were purchased from a commercial supplier (ChemBridge, Corp, San Diego, CA).
Binding of [125I]-G-CSF was measured using previously described method [25] with some modifications made in our lab [26]. [125I]-G-CSF (specific activity 46.55 TBq/mmol) was purchased from Perkin Elmer (Waltham, Massachusetts). Cells were incubated in 180 µL of binding medium containing 0.2% BSA, 5 mM MgSO4 and 50 mM Hepes, pH 7.2 at a concentration of 1.5 × 107 cells/ml and the appropriate concentration of [125I]-G-CSF. Incubations were carried out at room temperature with periodic shaking to ensure continuous mixing of cells and radioactive ligand. The incubation time of 2 h was estimated from preliminary experiments as being found sufficient to reach equilibrium. Nonspecific binding of [125I]-G-CSF was measured by incubations in the presence of a 100-fold molar excess of unlabeled G-CSF. At the end of the incubation, bound and free radio ligands were discriminated by using separating oil according to previously published method [27]. Namely, 80 µL aliquots were sampled from the incubation mixture, each aliquot was layered on 300 µL of separating oil placed in 500 µL polyethylene tubes and centrifuged for 5 min at 6000 rpm. The separating oil consisted of 1.5 parts dibutyl phthalate and 1 part bis(2- ethylhexyl)phthalate (Aldrich-Sigma, St. Louis, MO).
Bound and free ligand activities were counted after cutting tubes in two pieces and placing the tips and tops, respectively into separate scintillation vials. Counting was performed on Beckman Coulter LS6500 scintillation counter. Glacial acetic acid was employed to solubilize the pellet. The specific binding was determined from the amount of bound [125I]-G-CSF blocked by competition with excess unlabeled G-CSF. The parameters of saturation binding experiments including dissociation constant (Kd), maximum binding capacity (Bmax) and binding cooperativity (h) were calculated with GraphPad Prism 5 software (La Jolla, CA) by using nonlinear regression analysis.
Cells, prepared as described above, were incubated in 180 µL of binding medium containing 0.2% BSA, 5 mM MgSO4 and 50 mM Hepes, pH 7.2 at a concentration of 1.5 × 107 cells/ml and the appropriate concentration of [125I]-G-CSF. This was followed by addition of increasing concentrations of each study compound (10 to 3000 nM). After 2 hrs of incubation, the amount of bound and free [125I]-G-CSF was determined, as described above. From these data, the parameters of competition for the receptor were calculated with GraphPad Prism 5 software (La Jolla, CA). Each concentration was analyzed in triplicate and experiments were repeated three times. These experiments were conducted in human monocytic and neuronal cell lines.
THP-1 cells or SH-SY5Y cells 3 × 106 cells in 5 ml media in a 25 cm2 flask were treated with a) 100 ng/ml G-CSF, b) medium alone without G-CSF and c) three different concentrations of the test drug alone for 24 h. In addition, a set of flasks was co-incubated with the combination of G-CSF and 3 concentrations of test drug. Whole cell lysates (60 mg) were separated on 10% polyacrylamide gel electrophoresis-SDS (PAGE-SDS). Proteins were electrophoretically transferred to nitrocellulose membranes, blocked with 5% non-fat milk prepared on Tries buffered saline containing 0.1% Tween 20, washed and incubated with a polyclonal antibody against either Bcl2 (Cell Signaling) or PKCδVIII-specific polyclonal antibody (Patel laboratory) [28]. The housekeeping gene GAPDH was used as an internal standard. Following incubation with anti-rabbit IgG-HRP, gel images were digitally captured by ProteinSimple FluorChem™ and densitometric analysis was performed using AlphaView™ Software.
Densitometric results from Western blots were analyzed with twotailed t-tests when only two groups were compared. In other analyses where 3 groups were compared, one-way ANOVA followed by t-tests with Dunnett’s corrections for multiple comparisons was utilized. Experiments were performed at least three times; all data was presented as mean ± SEM. All statistical analysis of data was done via PRISM4 or PRISM5 statistical analysis software (GraphPad Prism, La Jolla, CA). All comparisons were considered significant at level of P<0.05.
In order to identify a mode of action for the Kusano compounds (Cmpds#1-3), a limited computational analysis was performed. In brief, the coordinates for G-CSF bound to its receptor were obtained from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/?term=1PGR). The coordinates showed a key point of interaction whereby a glutamate (E20) from G-CSF protrudes into a small pocket of GCSF-R, which was estimated to be a hydrogen bond accepting region. It was hypothesized that a molecule capable of interrupting this interaction could reduce binding of G-CSF with G-CSFR. Examining the Kusano compounds (Cmpds 1-3, Table 1) via simple docking, we found these 3 drugs were able to occupy this pocket with negative but high estimated free energies of binding. The energies did indicate that binding was possible. The occlusion of the binding site would impede the ability of G-CSF to interact with G-CSFR. The binding patterns suggest there are two key points of binding to take advantage of: two discrete binding sites at the proteinprotein interface and binding into a pocket to disrupt the interaction of Glu20 from GCSF with Tyr78 and Arg193 from GCSF-R. Based on this analysis, two additional compounds were synthesized at the Moffitt Drug Discovery Center (Cmpds 4, 5) and five more compounds were identified by screening chemical libraries (Cmpds 6-10) from ChemBridge Corp (San Diego, CA).
Increasing concentrations of radio-labeled G-CSF (alone or in the presence of 100 fold excess “cold” G-CSF) were added to the cell cultures to generate saturation curves. Specific binding of [125I]-G-CSF to receptors expressed in monocytes (THP-1 cells) revealed a Kd=57.49 pM and a Hill coefficient of 0.92. The Kd in the neuronal cell line was 607 pM with a Hill coefficient of 2.7 (Table 2).
| Monocytic (THP-1) Cells | Neuronal (SH-SY5Y) Cells | |
| Bmax | 901 pM | 4826 pM |
| Hill Coefficient | 1.19 | 2.71 |
| Kd | 53.8 pM | 608 pM |
Table 2: Specific Binding Parameters of [125I]-G-CSF to its receptor on THP-1 cells and SH-SY5Y Cells.
Binding of the ligand with G-CSF receptors expressed by monocytic cells revealed greater affinity (lower Kd) than binding to the neuronal cells. However, binding of the ligand receptors expressed by the neuronal line exhibits substantial “cooperativity”, indicated by Hill coefficient of 2.7 compared to 1.19 in the monocytic cell line. Cooperative binding refers to enhanced binding of ligand to a macromolecule when there are already other ligands present on the same macromolecule. The Hill coefficient describes the fraction of the receptor macromolecule saturated by ligand as a function of the ligand concentration; it is used to determine the degree of cooperative binding of the ligand to receptor. A coefficient of 1 indicates completely independent binding, regardless of how many additional ligands are already bound as demonstrated in the saturation curve of the monocytic cell line. Numbers greater than one indicate positive cooperativity, while numbers less than one indicate negative cooperativity.
The binding parameters, including dissociation constant (Kd), maximum binding capacity (Bmax) and the Hill Coefficient were calculated with GraphPad Prism 5 software using nonlinear regression analysis
A typical displacement study result is shown in figure 2 demonstrating the capacity of Cmpds 6 and 10 to displace [125I]-GCSF from its receptor on monocytic cells with an IC50 of 13.7 nM and 2.3 nM, respectively. The IC50 indicates the concentration at which the drug displaces 50% of the bound G-CSF from its receptor. The smaller the IC50, the greater is the receptor binding affinity. Cmpd 10 exhibited a six times greater binding affinity for the GCSF receptor than Cmpd 6. Four of the ten compounds tested were capable of significantly competing with G-CSF for binding on the receptor (Figure 3). Compounds 6 and 10, which displaced greater than 75% of the [125I-GCSF, were further studied for their capacity to mimic or block the intracellular signaling triggered by the natural ligand.
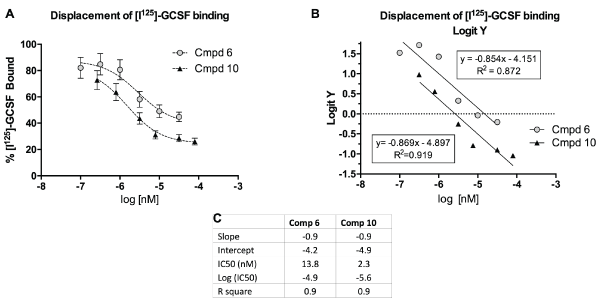
Figure 2: Compounds 6 and 10 displaced radio-labeled GCSF from receptor expressed on human monocytes.
A) Percent of radio-labeled GCSF bound to receptor (y-axis), expressed as mean ± SEM (n=6) plotted against increasing concentrations of Cmpd 6 and Cmpd 10. B) Logit Y (logistic regression analysis) vs concentrations of Cmpds 6 and 10 to calculate parameters of competitive binding of the drugs for the G-CSF receptor. C) Insert shows parameters of competitive binding for Cmpd 6 and Cmpd 10. The IC50 indicates the concentration of the drug that displaces 50% of the G-CSF from its receptor (smaller IC50=greater binding affinity for the receptor). Data based on 2 separate experiments with n=3 per experiment.
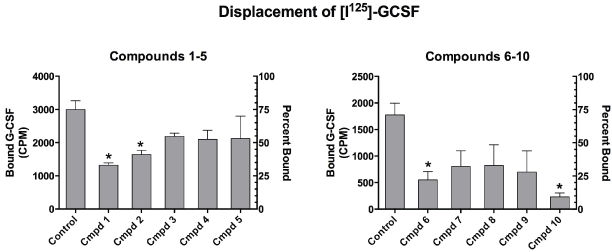
Figure 3: Displacement of [I125]-GCSF from its receptor expressed on monocytes by Compounds 1-10. The Y- axis on the left shows amount of bound G-CSF in CPM and the Y-axis on the right indicates percent of the radiolabeled GCSF bound to receptor.*Indicates significant displacement p<0.05 Cmpd 10 was the most effective, displacing 86.6% of I125-GCSF from its receptor.
Binding of G-CSF to its receptor triggers receptor homo-dimerization and activation of complex signal transduction and anti-apoptotic pathways which increase expression of STAT3, Bcl-2, and decrease expression of Bax [29-31]. Previous work in our laboratory had shown that G-CSF upregulates expression of PKCδVIII and Bcl2 in human monocytic and neuronal cell cultures [26]. Despite a lower binding affinity of G-CSF for receptors expressed by neurons compared to binding affinity in monocytic cells, we have reported that the expression of the anti-apoptotic protein Bcl2 and PKCδVIII, was greater at equivalent concentrations (100 ng/mL or 5.3 nM) in the human neuronal cell line than in the monocytic cells [26].
Comparison of the effects of G-CSF with the potential G-CSF mimetic drugs focused on the two most effective competitors for the G-CSF receptor, Cmpds 6 and 10. These drugs were evaluated for their capacity to activate or block the intracellular signaling triggered by G-CSF in human neuronal cells (SH-SY5Y) (Figure 4). Incubation of human neuronal cells with Cmpd 6 alone significantly increased Bcl2 protein expression at the low and intermediate, but not the high concentration (Figure 4C). In addition, Cmpd 6 alone stimulated PKCδVIII protein expression to levels equivalent to G-CSF alone. Incubation of the neuronal cells with both Cmpd 6 and G-CSF did not change the expression of either Bcl2 or PKCδVIII, indicating that Cmpd 6 did not act as an antagonist of G-CSF action in the neuronal cell line (Figure 4C). Incubation of the neuronal cell line with Cmpd 10 alone increased expression of both PKCδVIII and Bcl2 protein to levels equivalent to G-CSF alone (Figure 4D). In addition, Cmpd 10 at all three concentrations tested did not block the actions of G-CSF. To summarize both Cmpd 6 and Cmpd 10 appear to act as agonists at the G-CSF receptor while Cmpd 6 at the highest concentration increases PKCδVIII without changing Bcl2 expression.
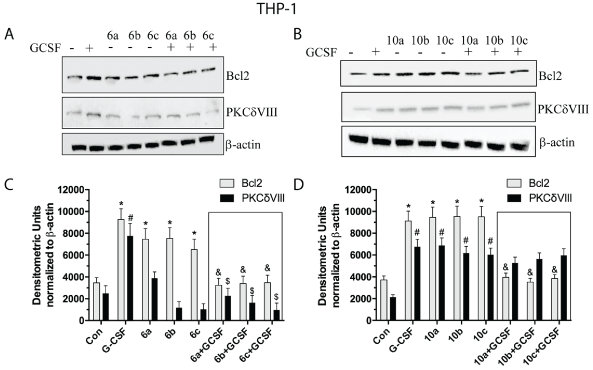
Figure 4: Western blots of PKCδVIII and Bcl2 expressed in SH-SY5Y neuronal cells.
(A) Immunoblot from cells incubated with three concentrations of Cmpd 6 alone and in the presence (or absence) of G-CSF (100 ng/mL). (B) Immunoblot from cells incubated 3 concentrations of Cmpd 10 alone and in the presence (or absence) of G-CSF (100 ng/mL). (C,D) Densitometric analyses of Immunoblots from A (Cmpd6) and B (Cmpd 10) respectively. Experiments were repeated three times. *=p<0.05 based on one-way ANOVA of the Bcl2 data followed by Dunnett’s multiple comparisons against vehicle control. #=p<0.05 based on one-way ANOVA of the PKCdVIII data alone followed by Dunnett’s multiple comparisons against vehicle control. Insert box shows effects of co-administration of G-CSF with Cmpds 6 or 10. $=p<0.05 based on one-way ANOVA followed by Dunnett’s multiple comparisons to G-CSF alone.
Studies in the monocytic THP-1 cell line revealed a different profile of drug-induced receptor activation (Figure 5). Cmpd 6 alone increased expression of Bcl2, but not PKCδVIII at all 3 concentrations. Unlike the results in the neuronal cell line, addition of Cmpd 6 to G-CSF blocked the increase in Bcl2 and PKCδVIII protein expression (Figure 5C). Hence, Cmpd 6 appears to act as a mixed agonist/antagonist in monocytic cell lines. Cmpd 10, at all 3 concentrations, was effective in significantly elevating expression of both Bcl2 and PKCδVIII (Figure 5D). Cmpd 10 also appeared to act as a mixed agonist/antagonist by blocking the effects of G-CSF on Bcl2 expression. However, Cmpd 10 did not antagonize the effects on PKCδVIII protein expression.
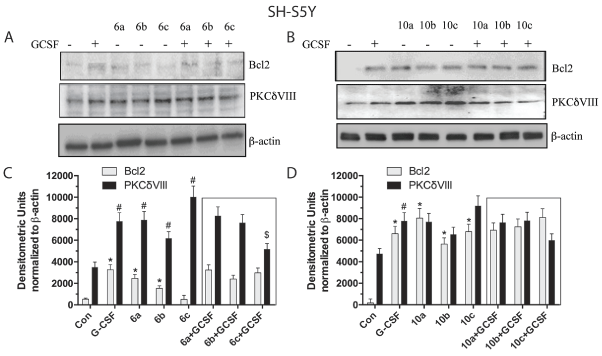
Figure 5: Western blots of PKCδVIII and Bcl2 expressed in THP-1 monocytic cells.
(A) Immunoblot from cells incubated with three concentrations of Cmpd 6 alone and in the presence (or absence) of G-CSF (100 ng/mL). (B) Immunoblot from cells incubated with 3 concentrations of Cmpd 10 alone and in the presence (or absence) of G-CSF (100 ng/mL). (C,D) Densitometric analyses of Immunoblots from A (Cmpd 6) and B (Cmpd 10), respectively. *=p<0.05 based on one-way ANOVA of the Bcl2 data followed by Dunnett’s correction for multiple comparisons against vehicle control. #=p<0.05) based on one-way ANOVA of the PKCdVIII data alone followed by Dunnett’s correction for multiple comparisons against vehicle control. Insert box shows effects of co-administration of G-CSF with Cmpds 6 or 10. &=p<0.05 based on one-way ANOVA of Bcl2 data followed by Dunnett’s correction for multiple comparisons to G-CSF alone. $=p<0.05 based on one-way ANOVA of the PKCdVIII data alone followed by Dunnett’s correction for multiple comparisons against G-CSF treatment.
The potential utility of G-CSF as a novel neurotrophic factor triggered a search for small molecules that interact with the G-CSF receptor to reproduce or block the biological actions of G-CSF. Production of small non-peptidergic molecules that mimic G-CSF effects on blood cell lineages had been previously undertaken in Japan [19] but the project was not pursued after 2004. Although the 3 compounds identified by Kusano cannot be obtained commercially, their reported chemical structures provided a practical starting point for the synthesis and search of drugs that interact with the G-CSF receptor [19].
Based on computer-based modeling of these 3 drugs and a search of drug libraries, we identified a total of ten compounds with potential to interact with the G-CSF receptor in human cell culture lines. Two of the 10 drugs (Cmpds 6 and 10) were shown to interact with the G- CSF receptor and to trigger signal transduction in human monocytic and neuronal human cell cultures. Cmpd 10 appeared to function as a G-CSF agonist in neuronal cell lines. Expression of both pro-survival proteins Bcl2 and PKCδVIII was increased by treatment with the drug in the neuronal cells. By contrast, Cmpd 10 at the lowest concentration antagonized the effects of G-CSF on the expression of Bcl2 and PKCδVIII in the monocytic cell lines. Therefore it may be possible to develop a combination drug (G-CSF+Cmpd 10) that will serve as a neurotrophic factor in vivo, free of potentially dangerous leukocytosis, thrombocytopenia and splenomegaly. This would be accomplished by stimulating neurons that bear G-CSF receptor (by both Cmpd 10 and G-CSF). At the same time Cmpd 10 would be capable of blocking the peripheral effects of G-CSF and thereby prevent excessive leukocytosis.
Although it is clear that Cmpds 6 and 10 can displace radio-labeled G-CSF from its receptors on both monocytic and neuronal cell lines, evidence for direct binding of these drugs to GCSF-R remains to be determined. This will require future experiments to assess direct drugGCSF-R binding using NMR (nuclear magnetic resonance) or other techniques. It will also be important to more fully investigate the earliest steps of intracellular signaling, such as receptor tyrosine phosphorylation or STAT3 phosphorylation and other intermediates leading to Bcl2 and PKCδVIII expression. The latter measures of biological effects of G-CSF receptor stimulation were emphasized because the clinical relevance of G-CSF as a neurotrophic factor involves the upregulation of the antiapoptotic factor Bcl2.
There are several disadvantages to the application of human G-CSF as a neurotrophic factor to treat neurologic diseases such as AD, PD, TBI and stroke: 1) It is produced by recombinant DNA technology and is therefore expensive to manufacture and costly to administer a course of treatment. 2) The primary peripheral actions of G-CSF (i.e. to stimulate hematopoiesis and increase circulating levels of neutrophils) limit the doses that can be used safely to treat brain disorders. 3) There are no specific G-CSF receptor antagonists capable of blocking the peripheral actions of G-CSF, leaving intact the direct neurotrophic effects in brain.
In conclusion, there is a need to develop drugs that mimic the neurotrophic and/or immune-modulating actions of G-CSF. The drugs are easier and cheaper to make than G-CSF. Structural modification of G-CSF mimetic drugs that selectively interact with central nervous system G-CSF receptors will lead to a new class of neurotrophic drugs, with minimal effects on leukopoiesis. Selective stimulation of central nervous system G- CSF receptors will also be beneficial for treating some CNS diseases in which excessive generation of leukocytes and thrombocytes is an unwanted side effect. In addition, discovery of G-CSF receptor antagonists impermeable to brain will be useful in distinguishing peripheral from central actions of G-CSF. G-CSF antagonists may also be therapeutic for blood diseases associated with over- activity of the G-CSF system.
Going forward the next series of experiments will determine whether in vivo administration of G-CSF mimetics will be useful in animal models of stroke, trauma and neurodegenerative disease. In addition, mechanistic studies will need to be performed to a) confirm binding sites using and b) determination of the earliest intracellular signals triggered by activation of the G-CSF receptor.
Supported by funds from the Helen Ellis Endowment to JSR.
JSR, VS, S. Song, and S. Sebti have submitted a patent application to develop G-CSF mimetic drugs as either replacements of G-CSF itself or as modifiers of G- CSF activity
Download Provisional PDF Here
Article Type: Research Article
Citation: Sava V, Song S, Kong X, El Bassit G, Patel N, et al. (2017) Small Molecules that Mimic or Antagonize Actions of Granulocyte ColonyStimulating Factor (G-CSF). J Drug Res Dev 3(2): doi http://dx.doi.org/10.16966/2470-1009.131
Copyright: © 2017 Sava V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access