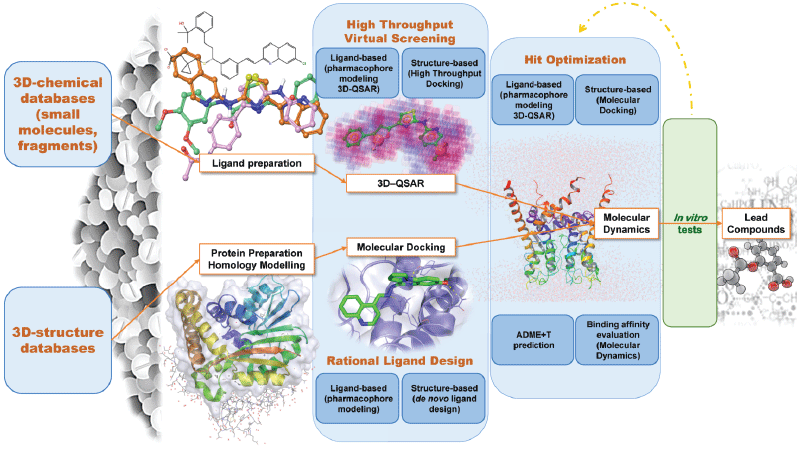
Figure 1. Computer assisted drug design (CADD) pipeline.

Giulia Chemi Simone Brogi*
European Research Centre for Drug Discovery and Development (NatSynDrugs) and Department of Biotechnology, Chemistry and Pharmacy, University of Siena, Via Aldo Moro 2, 53100 Siena, Italy*Corresponding author: Simone Brogi, European Research Centre for Drug Discovery and Development and Department of Biotechnology, Chemistry and Pharmacy, University of Siena, Via Aldo Moro 2, 53100 Siena, Italy, Tel: +39-0577-234389; E-mail: simonebrogi1976@hotmail.com; brogi32@unisi.it
In silico methodologies have become a pivotal part of the modern drug discovery process. Since their origin, computational techniques demonstrated to accelerate hit selection for a given drug target, and to significantly contribute to multiple stages of drug discovery (i.e. drug optimization) [1]. Accordingly, In silico drug design and discovery is in a state of constant and rapid development due to: (i) progress in the computer science which has led to the generation of powerful and affordable supercomputers, proliferation of available online tools, software and databases and development of more reliable algorithms; (ii) development of new experimental procedures for the characterization of biological targets (i.e. X-ray crystallography and NMR spectroscopy); (iii) the greater awareness of the molecular basis of drug action.
Herein we analyzed the most relevant computer aided drug design (CADD) breakthroughs. A variety of computational approaches with diverse potential applications along the drug discovery process (Figure 1) will be discussed and the last improvements of the In silico tools and methodologies examined.
Figure 1. Computer assisted drug design (CADD) pipeline.
Pharmacophore modeling, three-dimensional quantitative structureactivity relationships (3D-QSAR), Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) still remain the ligand-based (LB) methods of choice for fast virtual screening (VS) procedures. They are particularly powerful when the three-dimensional (3D) structure of the investigated protein is unknown [2-4]. VS is routinely employed by academia and pharmaceutical companies to identify novel chemical entities using public (e.g. ZINC database [5]), commercial or proprietary 3D-databases, with the possibility to screen billion of compounds in a short time, in order to reduce drug discovery costs [6]. The large amounts of available positive information (i.e. biological and structural data) allow the use of large dataset of known characterized compounds also for the development of 3D-QSAR models. These are crucial information for relating the structural and/or physicochemical properties of compounds to their activities in order to obtain more robust statistical In silico models for predicting activities of novel chemical entities [7]. CoMFA and CoMSIA are powerful tools to generate 3D-QSAR models to correlate the biological activity of a set of molecules and their 3D shape, electrostatic and hydrogen bonding characteristics. This correlation is derived from a series of superimposed conformations, one for each molecule in the set. The molecular fields around each conformation are calculated and the resulting 3D models can be used in VS protocols by using for example SYBYL-X Suite (Certara USA, Inc., Princeton, New Jersey, NJ).
Accordingly, the expertise in the generation of QSAR models and the development of statistical packages employing public available databases (considering theoretical or experimental data), made possible the realization of revised structure-relationships models. Below are reported important examples:
During the last decade, many scientific contributions appeared in the literature reporting improved QSAR methodologies. These advancements in structure-relationships models are extremely useful for rational drug design and for predicting ligands’ undesirable effects such as hERG K+ channel affinity. hERG K+ channel is a well-known antitarget responsible for cardiotoxic effects when targeted by centrally active drugs. In fact, the interaction of small molecules with hERG K+ channel is one of the major issues encountered by the pharmaceutical companies related to the drug development process. In the recent years several marketed drugs including astemizole, droperidol, terfenadine, lidolazine, sertindole, cisapride, and chlorpromazine have been withdrawn due to their relevant activity on hERG K+ channel. In this context, the generation of an adequate 3D-QSAR model based on hERG K+ channel blockers can assist the rational design of new potentially bioactive drugs devoid of hERG K+ channel affinity.
When the information of the 3D structure of the targets in complex with ligands are known, structure-based (SB) drug design approaches are useful for deriving SB pharmacophore models including excluded volumes (3D space portions in which the ligand cannot be located). The most commonly used software for generating SB pharmacophore models are: e-Pharmacophores, implemented in Maestro suite (Schrödinger, LLC, New York, NY), LigandScout (Inte: Ligand GmbH, Vienna, Austria), Catalyst, implemented in Discovery Studio (Accelrys, Inc., San Diego, CA, USA) and SB pharmacophore, implemented in Molecular Operating Environment (MOE) (Chemical Computing Group’s (CCG), Montreal, QC, Canada). Among them, the e-Pharmacophores method achieves the advantages of both ligand- and structure-based approaches by generating energetically optimized SB pharmacophores that can be used to rapidly screen billions of compounds. Indeed, SB models are employed in large-scale chemical databases screening procedures. As reported for LB methods, the progress in the experimental procedures and the recent improvements in CPU performances coupled to the availability of large public 3D-chemical libraries, gave a boost to this computational approach. Intriguingly, a relevant advancement in SB pharmacophore modeling is represented by the use of multiple SB pharmacophore models, built employing available crystal structures of the protein of interest in complex with diverse ligands, in VS protocols. The SB models can be used as sequential filtering tools for screening chemical libraries. Alternatively, they can be combined in an inclusive SB pharmacophore model taking into account the most relevant interactions of ligands into the receptor for generating a comprehensive SB pharmacophore [13]. In both ways, multiple SB pharmacophore models can be used in VS or in rational ligand design for identifying novel chemical entities or for optimizing existing hits. Likewise, LB and SB methods can be combined for obtaining more reliable hybrid computational protocols. Following this approach a performance increase in retrieving active molecules for a given target has been observed [2].
Regarding molecular docking techniques, important advances have been reported in the last few years relative to the In silico methods able to accommodate ligands into the binding site of their biological target Docking algorithms and scoring functions can generate structures of receptor-ligand complexes; they may rank compounds, and can estimate binding energies/affinities using specific algorithms. Consequently, molecular docking is the most commonly used tool to screen large chemical databases directly into the binding site of the selected biological target and can be applied to a wide array of different clinically-relevant proteins from human, parasites, viruses or other organisms. The abovementioned procedure, defined as High Throughput Docking (HTD), to date can be applied to a wide range of different targets [14,15]. This is possible thanks to the recent advances in computing capabilities, molecular simulation algorithms, the growing number of available experimental 3D protein structures, and of robust molecular models in turn produced by using novel homology modeling techniques (i.e. models generated by using multiple templates). Recently, molecular docking has also been applied in a novel way to identify and validate potential targets for active compounds (target fishing) [16]. Considering the great number of available crystal structures, paralleled by the advantages of phenotypic screening methodologies, HTD of bioactive compounds against relevant targets, coupled to the evaluation of the binding free energy, could aid the identification of an unknown target for a given bioactive compound. Classical docking programs such as Glide (Schrödinger, LLC, New York, NY), Autodock, Genetic Optimization for Ligand Docking (GOLD) (The Cambridge Crystallographic Data Centre, Cambridge, UK) can be used for target fishing procedure. Moreover an automated procedure, namely Virtual Screening Workflow (VSW), for performing multiple docking considering different proteins combined with the evaluation of the binding energy of the selected ligand has been implemented in Maestro suite (Schrödinger, LLC, New York, NY). Also, different online tools are available for identifying potential targets for a given small molecule (i.e SwissTargetPrediction [17]). These tools consider the similarity of the molecule without targets with compounds known to be active against specific ones. Despite the great improvement in amount and quality of data available in the Protein Data Bank (PDB), in terms of number of proteins, resolution of crystals, and in general in terms of reliability of the protein structures, several drawbacks for target fishing have been recently reported (i.e. improper pose predictions, scoring failures, binding site-ligand protonation interdependence, problems associated with generation of heterogeneous collection of binding cavities) [18]. Furthermore, modified molecular docking simulations aimed at improving the performance of standard molecular docking methods, continue to appear in the literature. A technique called “ensemble docking” was recently developed with the purpose of including protein flexibility in molecular docking calculation using multiple protein conformations. Consecutive docking calculations of each ligand into different conformations of a target receptor, represent a valuable method to mimic the dynamic nature of the biological target. In general, the performance of the “ensemble docking” technique is superior to that reported for docking into a single receptor conformation [19]. Another modified docking technique aimed at taking into account the protein flexibility is the Induced Fit Docking (IFD). This latter encompasses various steps such as ligands and proteins preparation and molecular docking, and induces conformational changes in the binding site to accommodate the ligand. In fact, this technique exhaustively identifies potential binding modes and related conformational changes by side-chain sampling, and backbone minimization in a selected radius around the poses found during the initial docking stage of the protocol [20]. Interestingly, a modified IFD protocol combining molecular docking with a rule-based approach to intrinsic reactivity has been developed for predicting potential sites of metabolism (soft spots) for a given ligand. This technique is useful for designing optimized derivatives which bear functionalities able to mask the identified soft spots. The IFD procedure calculates the accessibility degree of compounds to the cytochromes P450 (CYP) reactive center. The reactivity rules have been parametrized in P450 Site of Metabolism Prediction software (Schrödinger, LLC, New York, NY). The reactivity is predicted with a linear free energy approach based on the Hammett and Taft scheme, where the reactivity of a given atom is the sum of a baseline reactivity rate and a series of perturbations determined by the connectivity. This procedure is very useful for designing ligands with improved metabolic stability [21]. Further advances in docking calculations have been recently carried out by estimating ab initio charges of a given ligand for improving the docking predictions. The Quantum Mechanical-Polarized Ligand Docking (QPLD) workflow, implemented in Maestro suite (Schrödinger, LLC, New York, NY) [22], aims at improving the partial charges assigned to the atoms of the ligand in a docking run by replacing them with charges derived from Quantum Mechanical (QM) calculations. The computation is performed applying hybrid Quantum and Molecular Mechanical (QM/MM) method, where the protein is considered as the MM region and the ligand is defined as the QM region. In this way, the polarization of the charges on the ligand by the receptor is taken into consideration, and re-docking of the ligand is performed considering these QM charges. QPLD represents one of the recent applications of the hybrid QM/MM scoring method, which has rapidly become one of the most prevalent tools for investigating chemical reactivity in biomolecular systems, allowing the modeling of bondformation and -disruption [23]. However, the high computational costs for performing high-level QM calculations have restricted the applicability of these approaches. For hits identification by docking techniques, many improvements have been done about the scoring functions on the basis of entropy, desolvation effects, and target specificity
To investigate ligand-receptor complexes and in general the dynamics and thermodynamics of biological systems, Molecular Dynamics (MD) simulations represent one of the major computational resources, since their introduction in the late 70s [24,25]. MD procedure calculates the behavior of a molecular system in a considered time, providing extensive data on fluctuations and conformational changes of proteins and nucleic acids [26]. At the moment, several programs for performing MD simulation are available. Among them, ACEMD (Accelera Ltd, London, UK), Chemistry at Harvard Macromolecular Mechanics (CHARMM), Assisted Model Building with Energy Refinement (AMBER) (University of California, San Francisco, CA), Groningen Machine for Chemical Simulations (GROMACS), Nanoscale Molecular Dynamics (NAMD) (Theoretical and Computational Biophysics group, University of Illinois at Urbana-Champaign, Urbana, IL) and Desmond (D. E. Shaw Research, New York, NY), are the most popular. Currently, it is possible to simulate complex systems (whole proteins) in solution with an explicit solvent, membrane embedded proteins, or large macromolecular complexes like nucleosomes or ribosomes [27-29]. The improvement of the latter technique, in terms of the size of the investigated molecular systems as well as in terms of extent of the performed simulations (i.e. µs and/or ms of simulations) [30,31] is in large part a consequence of the use of high performance computing, parallelized computer architectures, and the accessibility to more efficient algorithms. The improvement of MD simulations is also linked to the development of more accurate force fields, able to evaluate in a detailed manner the system under investigation in order to reproduce the properties of every particle of that system [32,33]. Recent examples are the improvement of CHARMM, AutoDock4Zn and Optimized Potential for Liquid Simulations (OPLS) force fields. These advances mainly concern: (i) the improved accuracy in generating polypeptide backbone conformational ensembles for intrinsically disordered peptides and proteins (CHARMM) [34]; (ii) the inclusion of specialized potential describing the interactions of zinc-coordinating ligands, describing both the energetic and geometric components of the interaction (AutoDock4Zn) [35]; (iii) the addition of off-atom charge sites for representing halogen bonding and aryl nitrogen lone pairs and the complete refit of peptide dihedral parameters to better model the native structure of proteins (OPLS) [36]. The progresses in the development of more accurate force fields made possible a more accurate prediction of the binding free energy. This latter is extremely useful in the lead optimization step [37,38]. Despite the advances in MD simulations, the excessive computational cost in terms of time computing, very often discouraged scientists to run adequate number of replicas to assess the reproducibility of the approach. For bypassing the time-scale restrictions of conventional MD simulations, new hardware resources have been developed. Accordingly, MD simulations are currently performed by graphics-processing-units (GPUs), increasing the rate of calculation of an order of magnitude. Moreover, new processors for these MD simulations have been specifically designed, building supercomputers able to accomplish microseconds of simulation per day [39]. The lust to perform long simulations, within a realistic time, inspired the development of a variety of enhanced sampling practices, employing constraints to speed up the progression of a system. For instance, there are several methods such as metadynamics [40], accelerated MD [41], and Coarse-Grained MD (CGMD) [42] that alter the normal progression of the system with a history-dependent biasing potential along the trajectory followed by a properly selected set of collective variables. In CGMD the accessible timescales of MD simulations are increased and the actual degrees of freedom of the system are reduced by linking atoms into aggregate particles. Although this technique has proven useful to study biomolecular systems, it is plagued by reduced resolution since could not succeed in capturing subtle but relevant properties such as the H-bonds system in solvents. MD simulations can treat proteins and ligands in a flexible manner, allowing the relaxation of the binding site around the ligand considering the effect of explicit water molecules. More accurate MD-based methods are available for estimating the binding free energy (thermodynamic integration (TI), linear interaction energy (LIE), free energy perturbation (FEP), and molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA)). As above-mentioned, the accuracy in the estimation of the binding free energy using MD simulations can increase the efficiency of the drug discovery process [43,44]. A great improvement of the MD technique is represented by High Throughput Molecular Dynamics (HTMD), a novel technique based on the simulation throughput which allows understanding of drug interaction with biological targets with a high degree of resolution and accuracy. The method is a massive-scale MD simulation and can be used to screen chemical databases. This method showed, in the hit discovery step, higher performances than the HTD [45].
In summary, the huge technological progresses in hardware and software resources, algorithms design as well as the advances in the development of new experimental procedures for characterizing biological targets, make computer-assisted approaches (combined with specific biological investigations) the most valuable methods for limiting the time and costs of pre-clinical research. Furthermore, CADD approaches are employed for reducing the use of animals for in vivo testing, for helping the design of more effective and safer drugs and for contributing to the repositioning of known drugs. CADD represents a key instrument to assist medicinal chemists in drug design, discovery, development, and hit-optimization steps during the drug discovery process.
The authors wish to thank the European Research Centre for Drug Discovery and Development (NatSynDrugs) for the support. We are also grateful to Prof. Giuseppe Campiani, Prof. Stefania Butini, Prof. Sandra Gemma and Dr. Margherita Brindisi for their fruitful discussion during the elaboration of this manuscript. The British Society of Antimicrobial Chemotherapy (BSAC) is kindly acknowledged (grant number GA2016_087R to SB).
The authors declare no competing financial interest concerning the publication of this paper.
Download Provisional PDF Here
Article Type: Mini Review
Citation: Chemi G, Brogi S (2017) Breakthroughs in Computational Approaches for Drug Discovery. J Drug Res Dev 3(1): doi http://dx.doi.org/10.16966/2470-1009.129
Copyright: © 2017 Chemi G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access