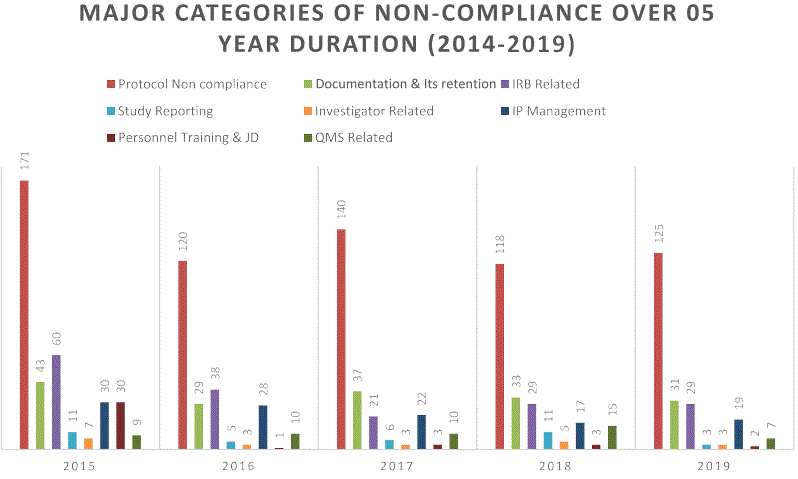
Figure 1: Major categories of non-compliances for clinical studies over five-years (2014-2019).

Vaibhav Choudhary*
Joint Director, Global Medical, Clinical and Regulatory Affairs, Haryana, India*Corresponding author: Vaibhav Choudhary, Joint Director, Global Medical, Clinical and Regulatory Affairs, Haryana, India, Tel: +91 9999200847; E-mail: Vaibhav.Choudhary@fresenius-kabi.com
Quality is a prerequisite during entire drug development cycle. Clinical studies are required to prove effectiveness of experiment drug and to evaluate its safety profile in humans. To have a smooth design and conduct of clinical studies; Quality-by-Design (QbD) approach can be integrated with Quality Management System (QMS) to ensure the end-to-end quality optimization. Quality by Design is not widely used currently in clinical research.
This manuscript proposes an effective integration of QbD with conventional QMS. We have applied the proposed integrated model in our clinical studies. We have studied elements of QbD thoroughly and identified Critical to Quality (CTQ) factors. These CTQ factors were integrated with QMS components to achieve improved quality. We found the integrated model to be pragmatic for clinical research and it has potential to change the Quality landscape in future.
QMS; QbD; Quality; CTQ factors; Clinical study; Data integrity; Clinical study design
Clinical studies involve evaluation of therapeutic effectiveness and safety of drugs, biological products, medical devices, and combination thereof in human volunteers. There is an increase in clinical studies due to the current pandemic. Due to revised guidance documents, the complexity of implementing studies has also increased. These changes create new challenges to clinical trial oversight, particularly increased variability in clinical investigator experience, site infrastructure, treatment choices, and standards of health care [1,2]. Thorough science and impeccable quality are absolute necessity throughout clinical study. The sponsors of clinical investigations are required to provide oversight to ensure adequate protection of the rights, welfare, and safety of human volunteers. [3-6].
Although Quality is a cornerstone for any clinical studies, there is also cost involved as a secondary but essential aspect for any organization. Now a days, outsourcing of clinical studies, either full or in part as well as technological advancements changed industry landscape. Globalization of clinical trials in terms of involvement of global stakeholders like CROs, clinical sites and multiple vendors increased the complexity of the clinical studies multifold. As it is associated with many ethical and scientific implications viz clinical study protocol compliance, monitoring, volunteer safety and rights, timely reporting, vendor qualification and management, and stakeholder communication [1,7]. Therefore, a change in quality program has become inevitable to safeguard safety of human volunteers and to ensure data integrity
The present approach fails to detect the real time assessment of ongoing clinical studies as it evaluates only certain aspects of clinical studies and not consider every stage of it from beginning to its end. In assuring and improving quality, a shift from current retrospective inspection-based quality management system to integrated model of Quality by Design in Quality Management is indispensable. This manuscript offers a framework of effective quality management by integration of Quality by Design (QbD) with conventional QMS that can be tailored to meet the requirements of clinical studies. The proposed integrated approach was successfully applied on conduct of a clinical study of a complex formulation.
The concept of Quality by Design (QbD) was first described by Juran JM in the early 1990s. In general, it defines quality as freedom from errors that matter. For a clinical trial, this could mean errors in study conduct or inaccuracies in data collection and/or reporting that affect the pre-specified study endpoints (and therefore harm study validity) or that jeopardize a patient’s rights or safety [8-11]. In order to ensure quality, it is important to understand nature of non-compliances detected by regulatory agencies. We have evaluated the database of FDA issued non-compliance observations in clinical studies to understand the trend [7,12]. The major categories of noncompliance over five-year duration (2015-2019) were formulated into a table (Table 1). For better understanding, the observations pattern was plotted into a bar-chart also (Figure 1).
Figure 1: Major categories of non-compliances for clinical studies over five-years (2014-2019).
| Year | Protocol non- compliance | Documentation and its retention | IRB Related | Study Reporting | Investigator Related | IP Management | Personnel Training & JD | QMS Related |
| 2015 | 171 | 43 | 60 | 11 | 7 | 30 | 30 | 9 |
| 2016 | 120 | 29 | 38 | 5 | 3 | 28 | 1 | 10 |
| 2017 | 140 | 37 | 21 | 6 | 3 | 22 | 3 | 10 |
| 2018 | 118 | 33 | 29 | 11 | 5 | 17 | 3 | 15 |
| 2019 | 125 | 31 | 29 | 3 | 3 | 19 | 2 | 7 |
| Source: Bioresearch Monitoring Observations (USFDA website) | ||||||||
Table 1: Major categories of non-compliances for clinical studies over five-years (2014-2019).
Quality by Design approach consists of defining quality goals at priori-to and develops the essential study documents, procedures to meet the pre-defined quality goals. QbD approach is intertwined with risk assessment and mitigation, hence designing a clinical study with QbD approach helps to eliminate potential risk(s). QbD approach can be divided into two main stages- Design stage and Execution stage [13]. Clinical trial quality ultimately rests on having a wellarticulated investigational plan with clearly defined objectives, study methodology, importance of ethics in clinical study and outcomes from the planned clinical study [9,13]. After design phase, protocols, Case Report Forms (CRF) and data collection procedures should be performed in a way, which support the defined design of clinical study. As depicted figure 2, Quality culture of an organization, especially trainings on ethics, data integrity, Good Document Practices are crucial. Moreover, transparent stakeholder communication is an integral QMS component throughout the clinical study, which includes any third-party collaborator (if applicable) as well.
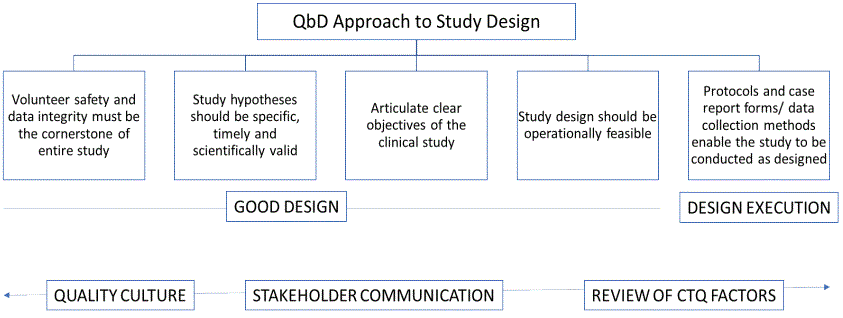
Figure 2: QbD approach- quality culture and transparent stakeholder communication.
Any variable critical to the outcome of the study or compromising the safety or rights of the study volunteers are called Critical Quality factors (CTQs) [13]. Sponsor organization should have an SOP in place for identification and documentation of Clinical to Quality Factors. All stakeholders involved in the clinical study are responsible to identify the critical to quality factors. The vital CTQ factors are depicted in figure 3. During study initiation phase, sponsors and investigators should identify the CTQ factors as per established SOP and document the same. This CTQ identification document shall be used further for developing clinical study essential documents. Based on data of 05 years USFDA observations [7] and our experience, we enlisted and elaborate the CTQ factors and their components (Table 2).
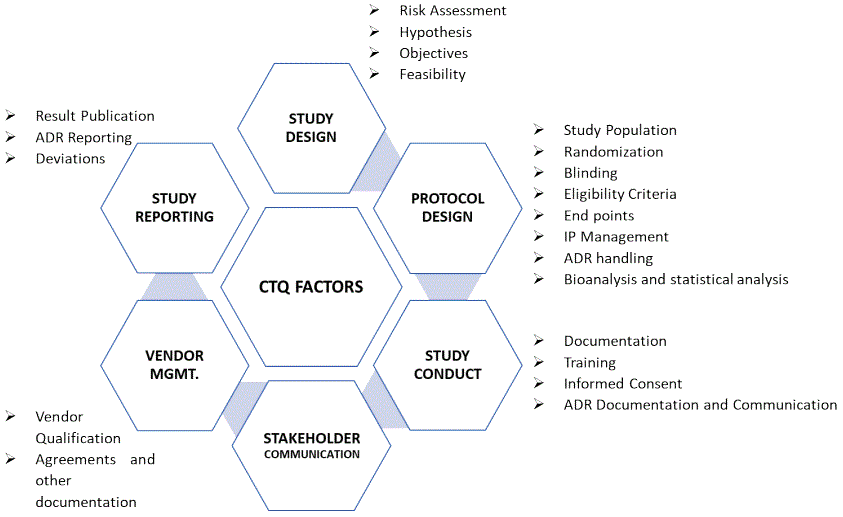
Figure 3: CTQ factors- key essential components for a clinical study.
| CTQ Factor | QbD Approach for Clinical Study |
| Study Design |
|
| Protocol Design |
|
| Study Conduct |
|
| Vendor Management | We utilized services only from vendors who had been previously qualified by our organization and had shown consistent compliance in services. Moreover, services were obtained only after robust agreements and service-related documentation with qualified vendors. |
| Communication to stakeholders |
We ensured the communication among stakeholders (including vendors) throughout the study conduct and complied with ICH GCP guidance also, in terms of communication to ethics committee, regulatory body. |
Table 2: CTQ factor and QbD approach for clinical study.
A proven QbD methodology used while study execution is the PlanDo-Check-Act (PDCA) cycle, to attain continuous improvement. PDCA cycle approach is effective to evaluate system/processes affecting CTQs [14], which is described below (Table 3).
| Plan: Recognize an opportunity and plan a change. |
| Do: Implement the plan, execute the process, collect the data, and measure the results. |
| Check: Review the test, analyze the results, and identify what you’ve learned. |
| Act: Act based on what you learned in the study step. If the change did not work, go through the cycle again with a different plan. If you were successful, incorporate what you learned from the test into wider changes. Use what you learned to plan new improvements, beginning the cycle again (Figure 4). |
Table 3: PDCA cycle approach.
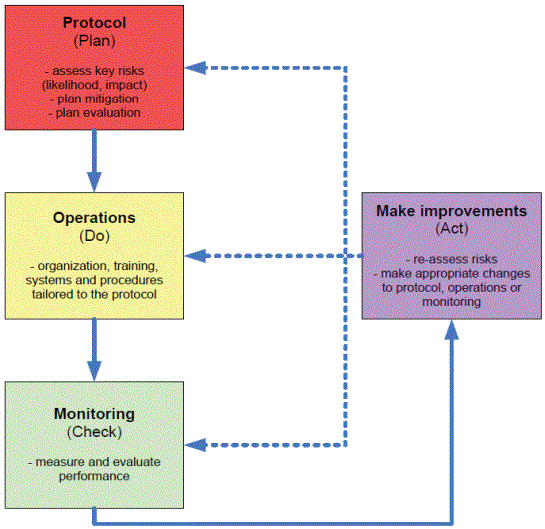
Figure 4: Plan-Do-Check-Act (PDCA) cycle.
Below is an example, which illustrates usage of PDCA cycle during a clinical study to ensure quality in the process.
It has been observed that minor documentation details were not completed by study staff personnel. The “plan” aspect of PDCA cycle involves asking questions to answer the root cause of an issue, such as-
• Who is responsible to ensure compliance of documentation?
• Whether there is a pattern in the incomplete documentation of forms?
• How the responsible personnel are trained and assessed before they involve independently in a clinical study?
• Whether other factors are involved for these documentation errors or not.
Once, the cause for an error is found after asking above questions, “Do” aspect determines the further course of action. Suppose in this case, the cause was found to be deficient training to responsible personnel and inadequate assessment, refresher training should be planned for responsible staff and he/she would be allowed to perform the task under observation. During “observation”, data would be collected for documentation compliance and error rate.
Then, “action” aspect would come into play. Actions would be performed, and compliance for documentation [9] shall be continuously monitored. For any non-compliance, CAPA would be taken and PDCA cycle would be repeated to ensure quality. The interval for PDCA cycle repetition is determined based on the design and needs of each clinical study.
Sponsor has the overall responsibility of the clinical study, although multiple sites are involved in a trial to ensure the validity of the data across the population, Sponsor is required to set up a best possible Quality Management System in place to ensure uniformity and integrity of the data. [ICH GCP E6(R2)] QbD integrated QMS ensures compliance in terms of effective documentation and optimized procedures during design and conduct of clinical study [15,16]. In this regard, we have identified critical QMS components, which may have direct or indirect impact on clinical study either in terms of data reliability and/or safety of clinical study volunteers (Table 4).
| Critical QMS Component | Advantage on clinical study conduct |
| Organizational policies and procedures | SOPs, Quality Directives, Policies and manuals decide quality framework for any organization. Such documents should be clear, concise, written in a language which can be understood by all study personnel in order to have a harmonized, smooth conduct of clinical study. |
| Training system | Training on regulatory guidance documents, data integrity aspects and necessary study documents ensure that study personnel perform the allotted task with thorough understanding and clarity of regulatory requirements. |
| Personnel roles and responsibilities | Clear role and responsibility allocation bring clarity for personnel to carry out work. Allocation of role and responsibility allows the user to get proper training before involvement in the study. Documentation of role and responsibility helps to reconstruct the study activity against personnel. |
| Quality Assurance audits | Appropriate sample size for auditing- either online or retrospective helps to ensure data reliability. Timely audit helps to resolve issues in timely manner, and scope of QA audit can be widened based on observations found. |
| Quality Control Unit | Thorough QC check and corrections help to resolve issues online and in general, QC check is performed for 100% study, which helps to remove any risks to data credibility. |
| Communication between stakeholders | Transparent communication and information flow help to operate a study without hindrance. Moreover, it also allows evaluation of study as an ongoing basis. |
| Human Factors (quality culture, personnel motivation) | Quality culture and an environment which encourages personnel to work motivated, ensures that responsible person contribute in best possible way to ensure quality. |
| Vendor/Supplier management | Evaluation of services provided any different vendors help to determine the right vendor for clinical study. Vendor Qualification prior to service agreement ensures desired quality throughout the study. |
| IT infrastructure and Computer System Validation | This factor assures that a computer-based resource used in the clinical study has been in compliance state in line with regulatory requirements. |
| Document management and Archive | Design of essential documents as well as pre-defined chain of custody for clinical study documents along with its archive in line with regulatory requirements ensure reconstruction of study at a later phase and retention of documents in unaltered manner. |
| Facilities and equipment | Facility design in such a way that different operations do not present any hindrance with each other and facility design should ensure that independence of critical operations like QA, Archive is achieved. Moreover, movement of material should not create any risk of contamination in case of laboratory operations. |
Table 4: Critical QMS components.
We conducted a clinical bioequivalence study considering the defined integrated quality management approach of QbD and QMS. The study was performed on an anti-cancer drug formulation which belongs to tyrosine kinase inhibitor. The study was executed considering the enlisted CTQ factors such as study design, protocol design, study conduct and vendor management, (Refer table 2) and critical QMS components (Refer table 4). There were many challenges during the beginning of clinical study such as very limited information about the pharmacokinetic characteristics of molecule, drug possess long plasma elimination half-life, and lack of precise information on intra- or inter-subject variability of drug. We assessed the initial risk of clinical study on selected drug considering the integrated QbD and QMS approach. Considering availability of very limited literature, a pilot study was planned in the initial phase of clinical development. The elements of QbD approach to study design depicted under figure 2 and the reported major categories of non-compliance (Table 1) were taken into consideration during clinical study design. The key information on drug pharmacokinetic behavior and intersubject variability were obtained from pilot study. Moreover, the major learning’s from execution of pilot study were implemented in pivotal study (viz IMP handling, record and administration; subject inclusion-exclusion criteria, ADR handling, training, stakeholder communication, bioanalysis, subject sensitization to compliance with study protocol and visit for ambulatory sample draw). The CTQ factors for pivotal study were further strengthen based on key learning’s and areas where we found the scope for further optimization (Table 2). Pharmacokinetic blood sample collection time point and duration were optimized to adequately characterize the maximum plasma concentration (Cmax) and Area Under Curve (AUC). Study sample size for pivotal study was computed based on the inter-subject variability obtained in pilot study and considering pivotal study power of at least 80%. The key advantages we observed through implementation of this integrated approach in our clinical study include:
» Initial risk assessment to determine CTQ factors provided better picture of clinical study and enabled stakeholders to evaluate all aspects in a holistic manner.
» Each milestone was achieved in the pre-defined timeframe which ultimately lead to effective conclusion of study in a timely manner.
» Due to clear communication channel, issues were resolved promptly.
» Personnel involved in the study did not deviate from the procedure defined in SOPs due to timely and thorough training beforehand.
» Our pivotal study was completed without any protocol deviation as study personnel were clear for assigned job and a transparent communication among different stakeholders.
» There were no regulatory observations found for this clinical study and we received authorization for the investigational drug.
» We did not face any issue while document reconciliation due to robust data management and pre-defined chain of custody for documents.
» We concluded a complex study with relative ease due to better study design and procedural controls in place.
In current manuscript, we enlisted the critical QMS components which ensured high quality standard across overall clinical development program (Table 4). The integrated approach of QbD into QMS was successfully implemented into a clinical study program to evaluate overall impact on quality and data integrity and subject safety. We concluded the clinical studies and found that QbD integrated QMS approach led to better data quality and compliance throughout the clinical study program. We humbly believe that this integrated approach is a scientific, risk-based, holistic and proactive approach to ensure safety of human volunteers and data integrity. We hope this article will initiate further discussions in the industry over this integrated approach for clinical studies.
The authors have no conflict of interests.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Choudhary V (2021) Design and Conduct of Clinical Studies by Integration of Quality by Design (QbD) into Quality Management System (QMS) - A Pragmatic Approach. Clin Res Open Access 7(2): dx.doi.org/10.16966/2469-6714.169
Copyright:© 2021 Choudhary V. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access