Introduction
Acute pancreatitis is common and is associated with substantial morbidity and mortality [1,2]. However, the mechanisms of disease pathogenesis are not well understood and, as a consequence, treatment options are limited. In the present study, we investigate regulation of nuclear transcription factor activation in exocrine pancreatic cells and test the hypothesis that Nuclear Factor-kappa B (NF-κB) and Activator Protein-1 (AP-1) modulate each other in exocrine pancreatic cells. Cooperative interactions between NF-κB and AP-1 have the potential to amplify pro-inflammatory responses to exocrine pancreatic cell stress and thus exacerbate acute pancreatitis.
NF-κB and AP-1 are ubiquitous transcription factors that regulate inflammatory and cell survival responses, are simultaneously activated by overlapping upstream kinases [e.g., Extracellular-Regulated Kinase (ERK)], and induce transcription of similar genes (Figure 1). Despite these close similarities, there are major differences in the regulation of NF-κB and AP-1 activation. In the quiescent state, the NF-κB subunits (e.g., p65, p50) reside in the cytoplasm complexed with Inhibitor of κB (IκB). IκB activation results in its own degradation with dissociation of the NF-κB/IκB complex, allowing nuclear translocation of NF-κB subunits and the ensuing gene transcription [3]. In contrast, AP-1 is a dimeric transcription factor protein group (e.g., Jun, Fos) located in the nucleus [4-6]. c-Jun forms homodimers that bind DNA and has a key role in AP-1-dependent transcription as c-Fos depends upon heterodimerization with c-Jun to bind DNA, while c-Fos on its own does not bind to the AP-1 DNA site [7].
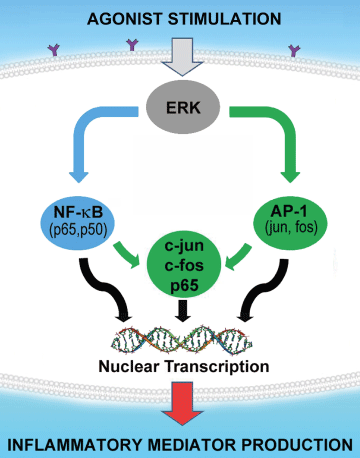
Figure 1: Hypothesis.
The central hypothesis tested in the present study is illustrated showing how nuclear transcription factors NF-κB and AP-1 are involved in cooperative interactions that increase gene transcription of pro-inflammatory mediators beyond the simple sum of their individual effects, thus resulting in synergistic augmentation of the acute inflammatory response. NF-κB subunit p65 and AP-1 component c-Jun promote each other’s transcriptional activity to amplify the summative response when both pathways are activated. The key component of the AP-1 complex of nuclear transcription factors is c-Jun as it forms c-fos/c-Jun heterodimers or c-Jun/c-Jun homodimers to induce transcriptional activity. As ERK MAPK regulates both NF-κB and AP-1, inhibition of ERK MAPK will potentially ameliorate acute pancreatitis.
Upstream of nuclear transcription factors, the Mitogen-Activated Protein Kinases (MAPK) such as ERK MAPK have received limited attention in exocrine pancreatic cell signal transduction [8-11]. We have previously shown that ERK MAPK regulates NF-κB-dependent gene transcription in exocrine pancreatic cells [12]. However, whether or not ERK MAPK regulates AP-1 in exocrine pancreatic cells has not previously been demonstrated. In this study, we demonstrate that expression of a Dominant Negative form of ERK (DN.ERK2) in exocrine pancreatic cells inhibits both NF-κB and AP-1 activation as well as both NF-κB-and AP-1-dependent gene transcriptions. We show that specific inhibition of NF-κB by expression of DN.IκBα diminishes AP-1-dependent transcriptional activity, while specific inhibition of the AP-1 pathway by expression of DN.c-Jun reduces NF-κB-dependent transcriptional activity. As these studies were performed using specific DNA binding site-responsive reporters, we conclude that NF-κB and AP-1 modulate each other in exocrine pancreatic cells. These findings underline the likelihood that cooperative interactions between NFκB and AP-1 exacerbate acute pancreatitis and that their upstream modulator ERK MAPK may be a target for therapeutic inhibition. These observations are in agreement with our recent finding that specific inhibition of ERK MAPK reduces mortality [13] in a mouse model of bile-pancreatic duct ligation-induced acute pancreatitis developed in our laboratory [14].
Materials and Methods
Experimental protocols were approved by the University of Iowa Institutional Animal Care and Use Committee (Protocol No: 1012246) and carried out according to Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, Bethesda, MD.
Antibodies
Antibodies were purchased from commercial sources: rabbit polyclonal antibodies against phosphorylated ERK 1/2 MAPK (Cat No: 9101), total ERK 1/2 MAPK (Cat No: 9102), total IκBα (Cat No: 4814) and phosphorylated c-Jun (Cat No: 9261), as well as rabbit monoclonal antibodies against phosphorylated NF-κB p65 (Cat No: 3039), total NFκB p65 (Cat No: 4764) and total c-Jun (Cat No: 9165) were from Cell Signaling, Danvers, MA. Other reagents included: mouse monoclonal antibody against TATA binding protein (Cat No: ab818; Abcam, Cambridge, MA); mouse monoclonal antibody against β-actin (Cat No: A5316; Sigma, St Louis, MO); goat anti-rabbit IgG-HRP (Cat No: sc2004; Santa Cruz Biotechnology, Santa Cruz, CA); goat anti-mouse IgGHRP (Cat No: 31438; Pierce Biotechnology, Rockford, IL).
AR42J cell culture
AR42J cells, a rat exocrine pancreatic malignant cell line (Cat No: CRL1492; ATCC, Manassas, VA), were grown in F-12K nutrient mixture Kaighn’s Modification (Cat No: 21127-022; Invitrogen, Grand Island, NY) with 20% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in poly-d-lysine coated culture dishes. Cells were incubated at 37°C in humidified 5% CO2. We have previously demonstrated close to 90% infection efficiency and excellent cell viability up to 48h after adenoviral infection of AR42J cells [15].
Recombinant adenoviral vector infection in-vitro
Replication-deficient recombinant adenoviruses expressing DN.IκBα (Ad.DN.IκBα, S32/36A mutation, Cat No: 1028), DN.c-Jun (TAM67, Cat No: 1046) or an AP-1-responsive luciferase reporter vector (Ad.AP-1.Luc, Cat No: 1670) were purchased from Vector Biolabs (Philadelphia, PA), those expressing an NF-κB-responsive luciferase reporter vector [16], Green Fluorescent Protein (Ad.GFP) or an empty vector control that does not express a transgene (Ad.EV) were made by the University of Iowa Vector Core Facility (Iowa City, IA), and that expressing DN.ERK2 (dual phosphorylation site T202/Y204 mutated to A202/F204) was purchased from Cell Biolabs, Inc. (San Diego, Calif., USA; Catalog No: ADV 113). Adenoviral infections and co-infections of cells were performed at an MOI of 5-10 Plaque Forming Units (PFU) per cell for 48 hrs at 37°C prior to stimulation for immunoblot analysis or luciferase assay.
Recombinant Adeno-Associated virus vector infection in-vivo and acinar cell isolation
We first transduced the mouse pancreas in vivo using the AdenoAssociated Viral (AAV) vector to express the NF-κB-responsive luciferase reporter, and then isolated the acinar cells and used only one adenoviral vector in vitro per well to perform the study. Compared to the adenovirus vector, the AAV vector has very negligible immunogenicity and is associated with undetectable inflammation [17-20]. Non-operated mice (male, 30-50 g, C57BL/6 from National Cancer Institute, Frederick, MD) were given AAV8.NF-κB.Luc 3e12 Viral Genome Particles (VGP) i.p. 2 wks prior to euthanasia. The pancreas was harvested fresh and primary cultures of acinar cells were isolated using methods described by the Williams group [21]. In brief, pooled pancreata were digested with collagenase in DMEM (containing 0.25% BSA, penicillin and streptomycin, and trypsin inhibitor), titrated, filtered through 150 µm mesh, and purified using a linear 4% BSA gradient. The isolated acinar cells were seeded in nontreated tissue culture plates at approximately 350-400 μg protein/ ml using 1 ml/well of DMEM, and then incubated at 37°C in a humidified atmosphere containing 5% CO2. The acinar cells were then infected with 5 MOI of Ad.EV, Ad.DN.IκBα, or Ad.DN.c-Jun. We have previously shown that the isolated acinar cells are viable, do not have excessive injury, respond appropriately with amylase release when stimulated, and that adenoviral transduction efficiency is excellent [22].
Lysate preparation and western blot analysis
For Western blots using whole cell protein extracts, AR42J or isolated acinar cells were lysed in modified RIPA buffer (50 mM TrisCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate) on ice for 10 min, following stimulation with cholecystokinin (CCK8, Cat No: C2175; Sigma, St. Louis, MO) or rat recombinant TNF-α (Cat No: 510-RT-010; R&D Systems, Minneapolis, MN). The soluble protein was collected following centrifugation at 15,000 xg for 10 min at 4°C. For Western blots using nuclear protein extracts, a commercial extraction kit (NE-PER Nuclear and Cytoplasmic Extraction Reagents, Cat No: 78833; Pierce Biotechnology, Rockford, IL) was used according to manufacturer’s instructions to compartmentalize nuclear and cytoplasmic proteins. Protein concentrations of whole cell homogenates and nuclear extracts were measured by Bradford protein assay (Cat No: 500-0006; Bio-Rad Laboratories, Hercules, CA) and lysates were denatured in SDS sample buffer and boiled for 3 min. Whole cell lysates (10-40 μg) or nuclear protein extracts (5- 10 µg) were subjected to SDS-PAGE on 12% gels and transferred to PVDF membranes. Membranes were probed with primary antibody against phospho-ERK 1/2, phospho-c-Jun, phospho-p65, p65 or IκBα, followed by either α-rabbit or α-mouse HRP-conjugated secondary antibody. Proteins were visualized using chemiluminescent detection (ECL Plus Western blotting detection reagent, Cat No: RPN2132; Amersham Pharmacia Biotech, Piscataway, NJ). Membranes were then stripped for 20 min at 50°C with a mild stripping buffer (69.3 mM SDS, 125 mM Tris pH 6.8, 243 mM β-mercaptoethanol) and re-probed for total ERK, total c-Jun, β-actin or TATA binding protein expression, as appropriate. Densitometric analysis of immunoblots was then performed using Image J software (Version 1.4, National Institutes of Health, Bethesda, MD).
Luciferase assays
AR42J cells were co-infected with Ad.EV (10 MOI), Ad.DN.ERK2 (10 MOI), Ad.DN.IκBα (5 MOI) or Ad.DN.c-Jun (10 MOI) in addition to 10 MOI of either Ad.NF-κB.Luc or Ad.AP-1.Luc for 48 hrs. Cells were harvested (Luciferase Cell Culture Lysis 5X Reagent, Cat No: E1531; Promega, Madison, WI) at 6 hrs following stimulation with TNF-α (10 ng/ml) or CCK (100 nM) and NF-κB- or AP-1-mediated gene transcription was evaluated by measuring luciferase activity (Luciferase Assay, Cat No: E1501; Promega, Madison, WI).
Statistical analysis
One-way ANOVA or paired t-tests were used (SigmaStat software, SPSS Inc., Chicago, IL). Three wells were studied in each group and results expressed as mean ± SEM. P values <0.05 were considered significant.
Results
Agonist stimulation activates ERK MAPK, NF-κB and AP-1 in AR42J cells
To determine the optimal stimulation time for ERK MAPK activation, AR42J cells were stimulated with TNF-α (100 ng/ml) or CCK8 (10 µM) for 1, 3, 5, 10 or 20 min prior to harvest. Immunoblots showed robust ERK MAPK phosphorylation (p-ERK) within 5 min of TNF-α stimulation, with a peak at 10 min, that returned to baseline levels by 20 min (Figure 2A). Strong ERK activation within 3 min after CCK stimulation peaked between 5 and 10 min and decreased by 20 min (Figure 2B). For time course studies of NF-κB activation, AR42J cells stimulated with TNF-α (10 ng/ml) for 10, 30, 60, 180 or 360 min showed nuclear translocation of NF-κB subunit p65 (increased phospho-p65 in the nuclear fraction) with an initial peak at 10 min that decreased by 30 min (Figure 2C). Activation then increased again from 1-3 hrs after stimulation before declining by 6 hrs (bi-phasic response). For time course studies of AP-1 activation, AR42J cells were stimulated with CCK8 (100 nM) for 15, 30, 45 or 60 min prior to harvest. AP-1 activation, as evidenced by phosphorylation of AP-1 protein c-Jun, peaked at 15 min following CCK stimulation and stayed elevated through 1h (Figure 2D).
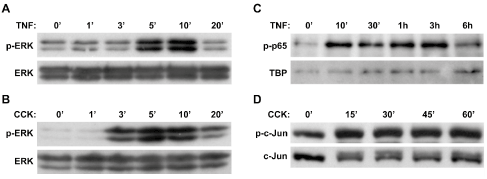
Figure 2: Activation of ERK, NF-κB (p65) and AP-1 (c-Jun) following agonist stimulation in AR42J cells. Immunoblot analysis showed increased ERK activation following stimulation of AR42J cells with either (A) 100 ng/ml TNF-α, or (B) 10 μM CCK, with phosphorylation of ERK (p-ERK) peaking at 5-10 min following stimulation with either agonist. Downstream of ERK activation, immunoblot analysis also showed (C) increased NF-κB p65 subunit activation (bi-phasic response) in the nuclear fraction following stimulation with 10 ng/ml TNF-α, as well as (D) increased activation of AP-1 component c-Jun in the total homogenate following stimulation with 100 nM CCK.
ERK regulates NF-κB and AP-1 in AR42J cells
Using Ad.DN.ERK2, we have previously shown that ERK regulates NF-κB-dependent gene transcription in AR42J cells [12]. Here, we confirmed this previous finding and also showed that ERK is involved in agonist-stimulated nuclear translocation of NF-κB subunit p65 and in c-Jun activation.
We first verified both the expression and functionality of the DN.ERK construct by infecting AR42J cells with Ad.EV or Ad.DN. ERK2 (5 MOI) for 48 hrs prior to stimulating the cells with CCK8 (100 nM) or TNF-α (10 ng/ml). ERK activation over baseline levels was seen in EV-infected cells following stimulation with either CCK (2.8- fold increase) or TNF-α (9.0- fold increase) for 10 min (Figure 3). Cells expressing DN.ERK showed diminished activation of ERK following stimulation with either CCK (71% decrease) or TNF-α (29% decrease) compared to stimulated EV-infected controls, while total ERK signal was much stronger in the DN.ERK expressing cells. These findings verify that DN.ERK was expressed and provide functional evidence of inhibition of ERK phosphorylation following agonist stimulation.
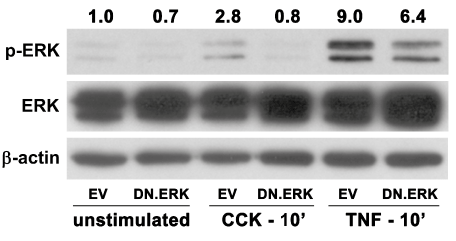
Figure 3: Expression of DN.ERK inhibits agonist-stimulated ERK activation in AR42J cells.
Cells were infected with 5 MOI of either Ad.EV or Ad.DN.ERK2 for 48hrs prior to stimulation ± 100 nM CCK or 10 ng/ml TNF-α for 10 min. ERK activation increased 2.8-fold following CCK stimulation and 9.0-fold following TNF-α stimulation of EV-infected control cells, while cells expressing DN.ERK showed diminished activation of ERK following CCK or TNF-α stimulation. β-actin expression was used as a protein loading control, while increased total ERK expression verified over expression of DN.ERK. Densitometric ratios of p-ERK to β-actin expression were normalized to unstimulated controls.
Strong NF-κB activation, evidenced by nuclear translocation of p65, was seen in TNF-α-stimulated EV-infected control cells after 20 min (Figure 4A and Figure 4B). DN.ERK-infected cells showed a 34% decrease in NF-κB activation (6.1-fold over baseline) following TNF-α stimulation, compared to EV-infected control cells (9.2-fold over baseline). Separately, AR42J cells were co-infected for 48hrs with 10 MOI of Ad.NF-κB.Luc and 10 MOI of either Ad.EV or Ad.DN.ERK2 prior to stimulation with TNF-α for 6hrs. EV-infected cells showed a 26.6-fold increase in transcriptional activity (as measured by luciferase activity) following TNF-α stimulation in comparison to unstimulated cells, which decreased by 47% in DN.ERK-infected cells. These results indicate that ERK regulates NF-κB activation and NF-κB-dependent gene transcription in AR42J cells.
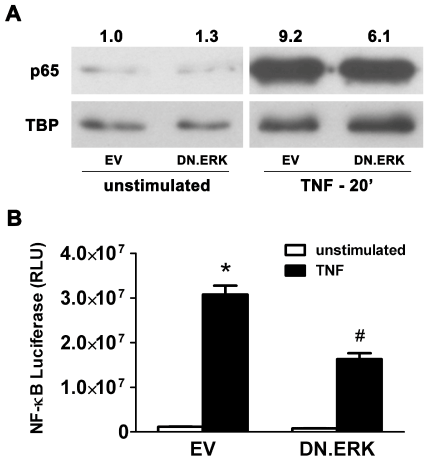
Figure 4: Expression of DN.ERK inhibits NF-κB activation and NF-κBdependent gene transcription in AR42J cells.
(A) Cells were infected with 5 MOI of either Ad.EV or Ad.DN.ERK2 for 48 hrs prior to stimulation with 10 ng/ml TNF-α for 20 min, followed by immunoblotting of the nuclear protein extract. Activation of NF-κB, evidenced by increased p65 in the nuclear fraction (nuclear translocation of p65), increased 9.2-fold following stimulation in EV-infected control cells, while stimulated DN.ERK-infected cells showed 34% decrease in NF-κB activation, compared to stimulated EV-infected control cells. TATA Binding Protein (TBP) expression was used as a nuclear protein loading control, and densitometric ratios of p65 to TBP expression were normalized to unstimulated EV-infected controls. (B) Cells were co-infected with 10 MOI of Ad.NF-κB.Luc and 10 MOI of either Ad.EV or Ad.DN.ERK2 for 48hrs prior to stimulation with 10 ng/ml TNF-α for 6hrs. Data are mean ± SEM; n=3 wells/group; ANOVA, asterisk (*) indicates significance vs. unstimulated control, pound sign (#) indicates significance vs. stimulated control, p<0.05; RLU= relative luciferase units.
To determine the effect of DN.ERK expression on AP-1 activation, AR42J cells were infected with 5 MOI of either Ad.EV or Ad.DN. ERK2 for 48 hrs followed by stimulation with 100 nM CCK8 for 10 min (Figure 5A and Figure 5B). Activation of AP-1 protein c-Jun was seen in EV-infected control cells following stimulation with phospho-c-Jun increasing 10.2-fold over unstimulated baseline levels in total cell lysate protein extracts. Cells expressing DN.ERK showed greatly inhibited activation [5.3-fold (48% decreases)] following CCK stimulation, compared to EV-infected controls. To test the effect of DN.ERK expression on AP-1-dependent gene transcription, AR42J cells were co-infected with 10 MOI of Ad.AP-1.Luc and 10 MOI of either Ad.EV or Ad.DN.ERK2 for 48 hrs prior to stimulation with 100 nM CCK for 6 hrs. EV-infected controls showed a 35-fold increase in transcriptional activity following CCK stimulation over unstimulated levels, which decreased by 75% in DN.ERK expressing cells. These findings indicate that ERK regulates activation of AP-1 component protein c-Jun and modulates AP-1-dependent gene transcription in AR42J cells.
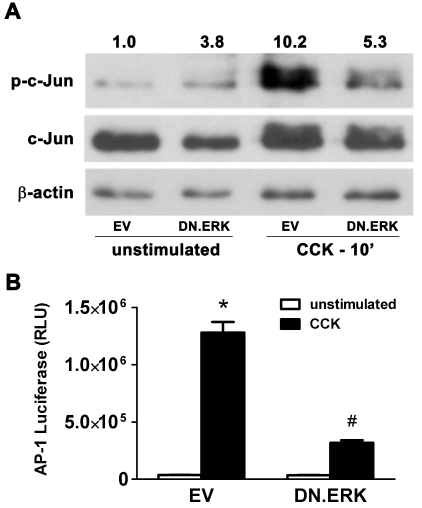
Figure 5: Expression of DN.ERK inhibits AP-1 activation and AP-1- dependent gene transcription in AR42J cells.
(A) Cells were infected with 5 MOI of either Ad.EV or Ad.DN.ERK2 for 48hrs prior to stimulation with 100 nM CCK for 10 min, followed by total protein extraction. Activation of AP-1 component c-Jun increased 10.2-fold following stimulation in EV-infected control cells, while stimulated DN.ERK-infected cells showed a 48% decrease in c-Jun activation, compared to stimulated EV-infected cells. β-actin expression was used as a protein loading control, and densitometric ratios of p-c-Jun to c-Jun expression were normalized to unstimulated EV-infected controls.
(B) Cells were co-infected with 10 MOI of Ad.AP-1.Luc and 10 MOI of either Ad.EV or Ad.DN.ERK2 for 48hrs prior to stimulation with 100 nM CCK for 6hrs. Data are mean ± SEM; n=3 wells/group; ANOVA, asterisk (*) indicates significance vs. unstimulated control, pound sign (#) indicates significance vs. stimulated control, p<0.05; RLU=Relative luciferase units.
NF-κB regulates c-Jun activation and AP-1-dependent gene transcription in AR42J cells
To evaluate NF-κB/AP-1 crosstalk, where NF-κB may regulate the AP-1 transcription factor complex, AR42J cells were infected with 5 MOI of either Ad.EV or Ad.DN.IκBα for 48 hrs and then stimulated with TNF-α (10 ng/ml) for 10 min. In stimulated EV-infected controls, c-Jun phosphorylation increased 3.0-fold over unstimulated levels, while stimulated DN.IκBα-infected cells showed only a 1.9-fold increase (Figure 6A). To determine if this inhibitory action extends to the transcriptional activity of the AP-1 complex, AR42J cells were co-infected with Ad.AP-1.Luc (10 MOI) and either Ad.DN.c-Jun (10 MOI) or Ad.DN.IκBα (5 MOI) for 48 hrs followed by stimulation with CCK8 (100 nM) for 6 hrs. As would be expected, stimulated EV infected controls showed a 57-fold increase in luciferase activity over unstimulated controls which decreased by 57% in DN.c-Jun infected cells (Figure 6B). However, CCK-stimulated increases in luciferase activity were reduced by 35% in DN.IκBα-infected cells indicating that NF-κB, in addition to AP-1 protein c-Jun, also plays a role in regulating AP-1-dependent transcriptional activity. The reduction in c-Jun activation and AP-1-dependent gene transcription following inhibition of the NF-κB pathway is evidence of crosstalk between the two key transcription factor pathways.
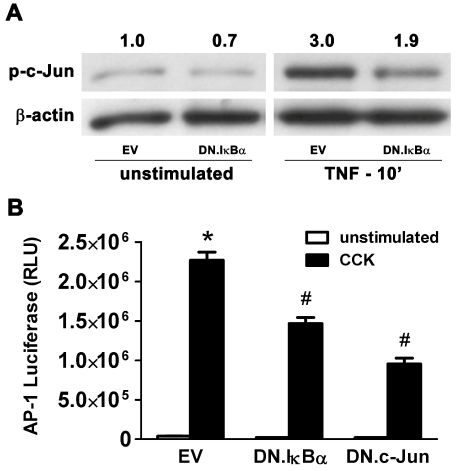
Figure 6: NF-κB/AP-1 Crosstalk: Expression of DN.IκBα inhibits activation of AP-1 component c-Jun and inhibits AP-1-dependent gene transcription in AR42J cells.
A) Cells were infected with 5 MOI of either Ad.EV or Ad.DN.IκBα for 48 hrs prior to stimulation with 10 ng/ml TNF-α for 10 min, followed by total protein extraction. Immunoblotting showed that activation of c-Jun increased 3.0-fold following stimulation in EV-infected control cells, while stimulated DN.IκBα-infected cells showed a 37% decrease in c-Jun activation, compared to stimulated EV-infected cells. β-actin expression was used as a protein loading control, and densitometric ratios of p-c-Jun to β-actin expression were normalized to unstimulated EV-infected controls. (B) Cells were coinfected with 10 MOI of Ad.AP-1.Luc and 10 MOI of Ad.EV, Ad.DN. IκBα or Ad.DN.c-Jun for 48hrs prior to stimulation with 100 nM CCK for 6 hrs.
EV-infected controls showed increased luciferase activity following stimulation, which decreased in stimulated DN.c-Jun expressing cells and even in stimulated DN.IκBα expressing cells. Data are mean ± SEM; n=3 wells/group; ANOVA, asterisk (*) indicates significance vs. unstimulated control (left bar), pound sign (#) indicates significance vs. stimulated control (right bar), p<0.05; RLU=Relative luciferase units.
AP-1 modulates NF-κB at the transcriptional level in AR42J cells
We then evaluated the converse circumstance where specific inhibition of the AP-1 pathway may also inhibit the NF-κB pathway. However, AP-1 components such as c-Jun would not be expected to modulate the initial activation of NF-κB subunits due to the nuclear location of AP-1 proteins and the cytosolic compartmentalization of the quiescent NF-κB/IκB complex. To verify this, AR42J cells were infected with 5 MOI of either EV or Ad.DN.c-Jun for 48 hrs followed by TNF-α stimulation (10 ng/ml) for 10 min. As predicted, TNFα-stimulated NF-κB activation (p65 translocation to the nuclear fraction) seen in EV-infected controls was not inhibited by Ad.DN.cJun infection (Figure 7A).
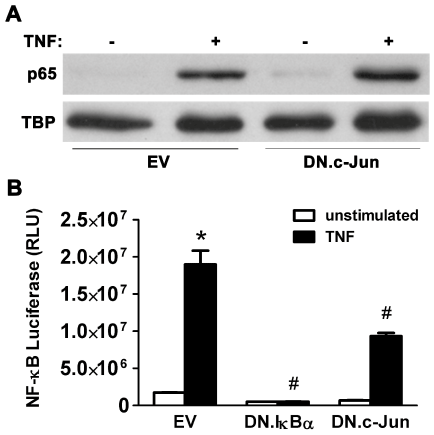
Figure 7: AP-1/NF-κB Crosstalk: Expression of DN.c-Jun inhibits NFκB-dependent gene transcription in AR42J cells.
(A) Cells were infected with 5 MOI of either Ad.EV or Ad.DN.c-Jun for 48hrs prior to stimulation with 10 ng/ml TNF-α for 10 min, followed by nuclear protein extraction. Immunoblotting showed that activation of p65 increased following stimulation in EV-infected control cells, and this activation was not inhibited by DN.c-Jun expression (as expected due to the nuclear location of AP-1 proteins and cytosolic location of inactive NF-κB proteins). Tata Binding Protein (TBP) expression was used as a nuclear loading control. (B) Cells were co-infected with 10 MOI of Ad.NF-κB.Luc and 10 MOI of either Ad.DN.IκBα or Ad.DN.c-Jun (Ad.EV in controls) for 48 hrs prior to stimulation with 10 ng/ml TNF-α for 6 hrs.
EV-infected control cells showed increased luciferase activity following stimulation, which decreased in stimulated DN.IκBα expressing cells and also in stimulated DN.c-Jun expressing cells. Data are mean ± SEM; n=3 wells/group; ANOVA, asterisk (*) indicates significance vs. unstimulated control, pound sign (#) indicates significance vs. stimulated control, p<0.05; RLU=Relative luciferase units.
To evaluate whether specific inhibition of AP-1 with DN.c-Jun expression affects NF-κB-dependent transcription, AR42J cells were incubated with 10 MOI of Ad.NF-κB.Luc and either 5 MOI of Ad.DN. IκBα or 10 MOI of Ad.DN.c-Jun for 48 hrs, followed by stimulation with TNF-α (10 ng/ml) for 6 hrs. In EV-infected controls, TNF-α stimulation led to a 10.9-fold increase in NF-κB transcriptional activity (Figure 7B). As expected, cells expressing DN.IκBα showed inhibition of TNF-α-stimulated increases in NF-κB transcriptional activity by 93%.
Interestingly, DN.c-Jun expression also led to a significant reduction (51%) in TNF-α-stimulated NF-κB transcriptional activity, indicating that AP-1 modulates NF-κB. Taken together with the data that NF-κB modulates activation of the AP-1 pathway, regulation of NF-κB transcriptional activity by AP-1 provides further evidence of crosstalk between the NF-κB and AP-1 pathways in AR42J cells.
NF-κB-dependent gene transcription in mouse isolated acinar cells
We then tested if DN.c-Jun modulates NF-κB transcriptional activity also in mouse isolated acinar cells. Non-operated C57BL/6 mice received AAV8.NF-κB.Luc 3e12 VGP i.p. 2 wks before the pancreas was harvested. Acinar cells were isolated and infected for 48 hrs with 5 MOI of either Ad.DN.IκBα or Ad.DN.c-Jun (EV in controls). Cells were stimulated with TNF-α 10 ng/ml for 6 hrs prior to luciferase assay. EV-infected control cells showed a 3.8-fold increase in luciferase activity following stimulation. As expected, expression of DN.IκBα inhibited TNF-α-stimulated increases in NF-κB transcriptional activity (by 83%, Figure 8). Confirming our findings in AR42J cells (Figure 7), expression of DN.c-Jun also reduced TNF-α stimulated NF-κB- transcriptional activity in isolated acinar cells (by 36%, Figure 8). This indicates that AP-1 also modulates the NF-κB pathway in mouse acinar cells, just as in AR42J cells.
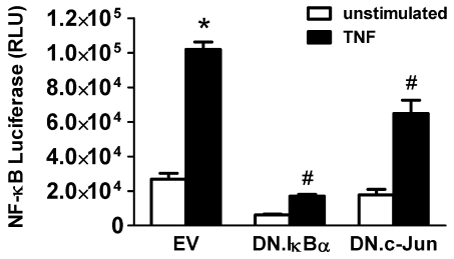
Figure 8: DN.c-Jun expression inhibits NF-κB-dependent gene transcription even in isolated acinar cells.
C57BL/6 mice were given AAV8.NF-κB.Luc (3e12 VGP) i.p. and acinar cells were isolated 2 weeks later. Cells were infected with 5 MOI of Ad.EV, Ad.DN.IκBα or Ad.DN.c-Jun for 48 hrs prior to stimulation ± 10 ng/ml TNF-± for 6 hrs. EV-infected control cells showed increased luciferase activity following stimulation, which decreased not only in stimulated DN.IκBα expressing cells but also in stimulated DN.c-Jun expressing cells. Data are mean ± SEM; n=3 wells/group; ANOVA, asterisk (*) indicates significance vs. unstimulated control, pound sign (#) indicates significance vs. stimulated control, p<0.05; RLU=Relative luciferase units.
Discussion
We present the important finding that cooperative interactions take place between the NF-κB and AP-1 nuclear transcription factor pathways in exocrine pancreatic cells. Using in vitro methods, we have shown that the NF-κB and AP-1 pathways modulate each other in exocrine pancreatic cells and that the upstream nitrogen-activated protein kinase ERK regulates both NF-κB and AP-1 pathways. The NFκB and AP-1 nuclear transcription factor pathways transmit signals for several cellular responses to changes in environment including acute inflammation, oxidative stress, infection, ionizing radiation, and mutagenic influences. NF-κB and AP-1 modulate genes involved in inflammation, immune regulation, cell growth and development, apoptosis and necrosis, and tissue injury and repair. The expression of a wide array of genes for pro-inflammatory mediators is regulated by NF-κB and AP-1 including cytokines, chemokines, oxygen-derived free radicals, adhesion molecules, and inducible effector enzymes [23- 25]. Binding sequences for NF-κB and AP-1 are present in exocrine pancreatic cell promoter regions of pro-inflammatory mediators and, therefore, NF-κB and AP-1 activation increase the expression of several mediators that promote acute pancreatic inflammation [23,26]. The regulation of the AP-1 pathway by NF-κB and the modulation of the NF-κB pathway by AP-1 imply synergistic augmentation of cellular signals within stressed exocrine pancreatic cells, thus increasing pro-inflammatory mediator production and worsening cell death responses in acute pancreatitis. The cooperative interactions between NF-κB and AP-1 are postulated to occur via a phenomenon known as cross-coupling that involves novel heteromeric complex formation at DNA binding sites where physical and functional interactions between NF-κB p65 and c-Jun/c-Fos lead to enhanced transcriptional activity at both κB and AP-1 enhancer-dependent promoters [4]. Such a crosstalk phenomenon between the key nuclear transcription factors NF-κB and AP-1 in pancreatic acinar cells conceivably contributes to the exacerbation of acute pancreatitis. Therefore, compounds such as the bioflavonoid curcumin that inhibit both NF-κB and AP-1, and are known to have beneficial effects in experimental models of acute pancreatitis [27], may have therapeutic roles in acute pancreatitis by also subduing synergistic interactions between the two pathways.
NF-κB and AP-1 have actions that are either pro-inflammatory or anti-inflammatory, depending on the upstream signal that activates them [3]. Our demonstration in the present study that ERK MAPK, a key stress kinase upstream of NF-κB and AP-1, modulates both nuclear transcription factor pathways in exocrine pancreatic cells is important in this context. Our results show that inhibition of ERK MAPK reduces activation of NF-κB and AP-1 and also diminishes NF-κB- and AP-1-dependent gene transcription in AR42J cells. Inhibition of ERK MAPK could potentially have beneficial effects in the management of acute pancreatitis by suppressing the individual effects of NF-κB and AP-1 activation, and also by subduing their cooperative interactions, thus providing another target for novel therapeutic initiatives. Interestingly, in a new model of duct ligationinduced acute pancreatitis in mice associated with substantial mortality developed recently in our laboratory [14,28], we have shown that specific inhibition of ERK MAPK significantly reduces mortality [13]. The improved survival with inhibition of ERK MAPK in this model suggests that ERK MAPK promotes pro-inflammatory-rather than anti-inflammatory- activation of NF-κB and AP-1 pathways.
In the present study, we performed time course studies of agonist-induced activation of ERK MAPK, NF-κB subunit p65, and AP-1 component c-Jun (Figure 2) to optimize conditions prior to experiments. We investigated the time course of CCK- and TNF-αstimulated activation of ERK MAPK, CCK-stimulated activation of c-Jun, and TNF-α-stimulated activation of p65 in AR42J cells. We have shown a bi-phasic pattern for TNF-α-stimulated nuclear translocation of NF-κB subunit p65, with an initial peak at 10 min and a second peak at 3hrs (Figure 2C). Evidence for two phases of NF-κB activation has been established in isolated acinar cells [29] and in other cell types [30,31]. IκB is composed of the subunits IκBα and IκBβ which maintain NF-κB in its quiescent form in the cytosol by masking its nuclear localization sequence. IκBα is rapidly but transiently degraded and believed to be responsible for the first phase, while IκBβ degrades slowly and is thought to be responsible for second phase of NF-κB activation. In the present study, we have verified that the dual phase of NF-κB activation occurs in AR42J cells just as is seen in isolated acinar cells [29], hepatocytes [30], and skeletal muscle cells [31].
AR42J cells offer many advantages in the study of signaling mechanisms in the exocrine pancreas such as the ease of cell culture maintenance, lower cost compared to pancreatic harvest from rodents, high transduction efficiency with adenoviral vectors, good tolerance of adenoviral infection, and the endogenous expression of receptors for many agonists (e.g., CCK, TNF-α)[15,26,32,33]. It can be practical to perform pilot studies in AR42J cells before confirming the salient findings in isolated acinar cells. Here, we performed a series of studies relevant to the central hypothesis in AR42J cells and, after obtaining encouraging results, confirmed that AP-1 modulates NF-κBdependent transcription even in mouse acinar cells. A novel approach using the AAV vector for in vivo expression of NF-κB.Luc prior to in vitro studies of mouse acinar cells allowed us to avoid the potential acinar cell injury with in vivo use of adenoviral vectors or in vitro use of more than one adenoviral vector. Confirmation of the converse situation, that NF-κB modulates AP-1-dependent gene transcription even in mouse acinar cells, was not studied as the AAV8.AP-1-Luc reagent has not yet been generated by us.
CCK and TNF-α are known activators of nuclear transcription factors and are often used in vitro to investigate pro-inflammatory pathways in exocrine pancreatic cells [15,22,26,27,32-39]. Using EMSA with super shift, we showed the p65 subunit of NF-κB is activated in AR42J cells stimulated by TNF-α [15]. In AR42J cells stimulated by CCK-analog cerulein, one study showed that NF-κB regulates IL-6 production [32] and another study showed that NFκB and AP-1 regulate IL-8 production [33]. Studies in isolated acinar cells showed that NF-κB regulates chemokine (MOB-1, MCP-1) and cytokine (IL-6, TNF-α) expression following stimulation with CCK [26,38]. Interestingly, complete suppression of MIP-2 up regulation in isolated acinar cells required dual inhibition of NF-κB and AP-1 [40], supporting the hypothesis studied here.
In mice, we developed a model of distal bile-pancreatic duct ligation-induced acute pancreatitis that is associated with systemic inflammation, multi-organ dysfunction, and a high mortality rate [14,28]. In this mouse model, we showed activation of ERK MAPK, NF-κB (p65), and AP-1 (c-Jun) in the pancreas, activation of ERK MAPK in the pancreas and distant organs, and elevated cytokine concentrations in the pancreas and lung [14,28,39,41]. In rats, EMSA has shown that pancreatic NF-κB activation occurs within 1hr and is increased at 24hrs with ligation-induced acute pancreatitis; gel super shift assay showed that the antibody to p65 subunit shifted the NF-κB band whereas anti-p50 antibody did not [42]. Pancreatic activation of NF-κB and AP-1 has also been shown in rats with acute pancreatitis induced by intra-duct injection of taurocholate [37] and that induced by cerulein or CCK plus ethanol diet [27].
In animal studies, inhibition of NF-κB improves survival in experimental acute pancreatitis [25,26], while over expression of NF-κB in the pancreas in vivo is associated with pancreatic and systemic injury [24]. N-Acetyl-L-Cysteine (NAC) and Pyrrolidine Dithiocarbamate (PDTC) are antioxidants that are non-specific inhibitors of NF-κB with beneficial effects in several animal models of pancreatitis [43-48]. Cerulein-pancreatitis was less severe in transgenic mice with knock out of NF-κB/p105 or mutation of the IKK2 gene [49,50]. Curcumin ameliorates acute pancreatitis and inhibits activation of both NFκB and AP-1 in two rat models of acute pancreatitis in parallel with inhibiting the induction of mRNA for several inhibitory mediators [27]. Many other studies showing beneficial effects of NF-κB inhibition have been reported and are well summarized in a recent review [26]. NF-κB activation in extra-pancreatic sites such as the lung, liver, and endothelium has also been described in animal models [26].
Evidence for NF-κB activation in human acute pancreatitis and systemic inflammation is available but limited. Significantly increased NF-κB activation in peripheral blood mononuclear cells of acute pancreatitis patients was observed within 24 hrs of symptom onset [51,52]. In clinical investigations of sepsis and septic shock, NF-κB is implicated in the development of acute lung injury [53]. Alveolar macrophages in patients with acute respiratory distress syndrome show NF-κB activation [54]. The present study underlines the need for more investigations into the role of NF-κB and AP-1 in experimental and human acute pancreatitis and the potential beneficial effects of inhibiting these key regulatory pathways that cooperatively exacerbate acute pancreatitis. Previous reports have underlined the functional roles played by NF-κB and AP-1 in cancer and other diseases [55- 57] and the potential for using them as targets for therapy [58-61]. A weakness of our study is that luciferase assays using the dominant negative forms of NF-κB inhibitor protein IκBα (DN.IκBα) and of AP-1 component protein c-Jun (DN.c-Jun) might not clearly depict acute inflammatory responses in exocrine pancreatic cells while attempting to elucidate the pathogenesis of acute pancreatitis. For instance, a recent study has shown epigenetic regulation of early- and late-response genes in acute pancreatitis [62].
Conclusion
The NF-κB and AP-1 pathways modulate each other in exocrine pancreatic cells. ERK MAPK, which regulates both NF-κB- and AP1-dependent gene transcriptions, is a potential target for therapeutic inhibition in acute pancreatitis.
Author Contributions
Twait EC performed the basic science experiments and acquired the data; Samuel I conceived the central study hypothesis; Twait EC and Samuel I designed the experiments, analyzed and interpreted the results and data, and wrote the manuscript. Supported by research grant awards to Samuel I: (a) Grant R01 DK-071731, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH), Bethesda, MD; (b) American Recovery and Reinvestment Act-Supplemental Award to NIH R01 DK-071731.
Institutional Review Board Statement
Not applicable (no human subjects)
Institutional Animal Care and Use Committee Statement
Experimental protocols were approved by the University of Iowa Institutional Animal Care and Use Committee (Protocol No. 1012246) and carried out according to Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, Bethesda, MD.
Conflict-of-Interest Statement
The authors do not have any conflicting interests to disclose.
Data Sharing Statement
No additional data are available.