Introduction
Vitiligo is an acquired hypopigmentary disease resulting from the loss or dysfunction of epidermal melanocytes. The understanding of its etiology is incomplete, yet various studies affirm implications of cellular cytotoxicity, oxidative stress, and systemic immune dysregulation. Epidermal melanocytes are innately susceptible to excessive free radicals and consequent oxidative stress compared to the other skin cell types [1-3]. Because melanin synthesis involves the generation of Reactive Oxygen Species (ROS) and melanin intermediates, melanocytes maintain an inherent pro-oxidation; this vulnerable state in combination with exposure to aggravating factors like Ultraviolet Radiation (UVR), hormones, stress, or cytotoxic compounds can lead to the progression of both dyspigmentation or oxidative driven skin diseases [4-7]. Thus, effectively managing pigmentary disorders while simultaneously encouraging melanocyte vitality is of consequence.
The transcription factor nuclear factor, erythroid 2-like 2 (Nrf2) is considered a central regulator for cell protection and survival through binding to the Antioxidant Response Element (ARE) present in the promoter of major antioxidant enzymes. The Nrf2-ARE pathway is stimulated by changes in the redox state of the cell and functions to restore homeostasis by upregulating antioxidant, xenobiotic metabolizing, and other cytoprotective proteins and enzymes constituting over 600 gene targets [8-9]. Scientific literature is also progressively uncovering a substantial role for Nrf2 in the metabolism and protection of skin cells [10-13].
Most research links redox imbalance to the pathogenesis of vitiligo [14-16]. Redox implications including systemic increases in oxidant status with simultaneous decreases in antioxidant status; downregulation of enzymatic antioxidants like Catalase (CAT), Glutathione Peroxidase (GPX), and Glucose-6-phosphate Dehydrogenase (G6PD); and upregulation of ROS Hydrogen Peroxide (H2O2) and Peroxynitrite (ONOO-) have been documented [17-25]. Various scientific studies associate vitiligo with multiple classical manifestations of oxidative stress such as lipid peroxidation, impaired signaling, structural irregularities, and dysfunction of cellular organelles [26-31]. Intriguingly, recent studies indicate impaired Nrf2-ARE pathway signaling and functional elements in vitiligo pathophysiology [32-35]. Thus, normalizing the cellular redox status and increasing protection from excessive ROS buildup potentially through selective induction of Nrf2-ARE pathway is a prime strategy for preventing the aggression of the oxidative driven diseases like vitiligo.
Galactomyces ferment filtrate, commercially known as Pitera™, is currently used as a cosmetic ingredient in skincare. GFF is a yeast derived extract that contains a unique composition of vitamins, minerals, small peptides, and oligosaccharides. Studies with GFF elucidate anti-aging, barrier health, and putative hypopigmenting effects on skin [36-40]. However, the full characterization of its properties is still in progress.
In this study, we investigated the treatment of GFF in normal melanocytes to further understand its antioxidant capacity and cellular health inducing benefits. We analyzed this at various time points, assessing the genomic, protein, and physiologic sequential consequences of GFF. We demonstrated that GFF treatment neutralizes potential oxidative assaults and upregulates downstream phase II antioxidant enzymes through activation of the Nrf2-ARE pathway in melanocytes. Such properties maybe useful for melanocyte redox pathologies including vitiligo.
Materials and Methods
Cell culture
Primary cultures of Normal Human Epidermal Melanocytes (NHEM) were established from discarded neonatal foreskin obtained from different skin types as previously described [41]. Tissues were procured from University Hospital in Cincinnati or from the Christ Hospital in Cincinnati. Patient consent was not required for experimentation because of USA laws regarding left over human tissues from surgery. Briefly, foreskins were washed with betadine and Phosphate Buffered Saline (PBS), sectioned and incubated in 0.25% trypsin, then rocked at 4°C overnight. Tissues were vortexed and centrifuged to separate the epidermis and dermis. The epidermal fraction was plated in T25 or T75 flasks with melanocyte growth medium. Melanocyte growth medium consisted of MCDB-153 (Sigma-Aldrich) supplemented with 4% Fetal Bovine Serum (FBS), 1% antibiotic/antimycotic, 15 μg/mL Bovine Pituitary Extract (BPE), 5 μg/mL insulin, 8 nM 12-O-Tetradecanoyl-Phorbol-13-Acetate (TPA), and 0.6 ng/mL Basic Fibroblast Growth Factor (bFGF) to aid in cell proliferation, dendricity, and melanization. Cultures were maintained in a humidified incubator with 5% CO2 at 37°C. Growth medium was changed every three to four days. For experimentation, moderately pigmented NHEM assessed for purity, by visible inspection of morphology and pigmentation, were selected and used from passages 3-5. Subsequent assessments for quality control were performed by Western blot analysis for melanogenic proteins expression, Tyrosinase (TYR) and/or Tyrosinase Related Protein 1 (TYRP1), and by immunocytochemistry for pankeratin expression.
GFF treatment
GFF was prepared using two different proprietary filtration methods: a routine method GFF-A, and an enhanced method GFF-B. Filtrates were prepared at Procter & Gamble Innovation Godo Kaisha, Kobe Technical Center; shipped to the laboratory on dry ice; and stored at 4°C. NHEM cultures were plated and incubated in experimental growth medium consisting of MCDB-153 supplemented with 6% FBS, 1% antibiotic/antimycotic, 15 μg/mL BPE, 5 μg/mL insulin, 2 nM TPA, and 0.15 ng/mL bFGF for 48 hours before the start of experiments. During experimentation, NHEM were treated with 0-10% GFF-A or GFF-B added to the experimental growth medium every 48 hours for 5-7 days. In single day experiments, NHEM were administered a single dose of 0-10% GFF-A or GFF-B added to their experimental growth medium and then harvested at the indicated time point. All cultures were maintained in a humidified incubator with 5% CO2 at 37°C until experiment harvest.
4TBP administration
4-Tertiary Butylphenol (4TBP) was administered to NHEM as previously described [42]. In short, 4TBP (Sigma-Aldrich) was dissolved in 70% ethanol and added to the experimental growth medium at indicated final concentrations (0-300 μM). Control NHEM were treated with 70% ethanol vehicle (maximum of 0.5% final concentration of alcohol).
UV irradiation
Caucasian/lightly-pigmented NHEM were incubated in PBS and irradiated with UVB 90 or 105 mJ/cm2 using FS20 lamps, with a peak emission at 313 nm, as previously described [43]. After irradiation, cells were incubated with 0-10% GFF-B in experimental growth medium until indicated harvest.
MTT cell viability assay
NHEM were treated with 200 μM 4TBP and 0-10% GFF-B, alone or in combination, for 6 days. Upon harvest, cell viability was measured by MTT assay (Bio Assay Systems) according to the manufacturer’s instructions. In brief, 1 × 104 cells/well was seeded in triplicate in a 6-well plate to attach overnight. MTT reagent (tetrazole) was added to the cells and incubated in a 37°C humidified chamber for 4 hours. The tetrazole is converted to formazan in mitochondria of living cells. The formazan crystals formed were solubilized in solubilization buffer and the wavelength was read in a microplate reader (Bio-Rad, 550). Fixation was used as a negative control. Cell viability was calculated from the absorbance readout and the results are expressed as a percentage of the control, n=3.
Fluorescent ROS visualization
NHEM were challenged with 300 μM 4TBP alone or in combination with 10% GFF-B or 30 U Catalase (Sigma-Aldrich) for 90 minutes. ROS generation was visualized using Image-IT® LIVE Green Reactive Oxygen Species Detection Kit (Molecular Probes) according to the manufacturer’s instructions as previously described [42]. Images in both phase-contrast and fluorescence microscopy were captured on Olympus IMT-2 inverted microscope (20X). To quantitate ROS, multiple images per group were size standardized and measured for mean intensity (minus background) using Image J to produce corrected total area fluorescence (CTAF), n=30.
H2O2 generation luminometer assay
Caucasian/lightly pigmented NHEM were treated with or without 10% GFF-B in PBS and immediately irradiated with UVB (105 mJ/cm2 ) as mentioned above. Samples were harvested at various timepoints (0, 15, 30, and 45 minutes) and then analyzed for H2O2 generation by luminescence of luminol as previously described [44]. In short, aliquots of PBS were transferred to tubes with respiratory buffer and luminol (Sigma-Aldrich). H2O2 release was determined by luminescence measured with a luminometer (Turner, TD10e). Readings were plotted against a standard curve with known H2 O2 concentrations. Results are expressed as pMol H2O2 / 1 × 105 cells, n=3.
RNA-seq transcriptome profiling
Illumina HiSeq-based next generation sequencing RNA-seq analysis was procured from the Genomics, Epigenomics and Sequencing Core (GESC), Department of Environmental Health, University of Cincinnati College of Medicine. A Caucasian/lightly pigmented NHEM cell line with a functional MC1R (determined by cyclic AMP analysis performed by the Dr. Zalfa Abdel-Malek laboratory) was treated with 10 % GFF-B. Large samples (plated at 4 × 106 cells) were harvested at two timepoints: 24 and 120 hours (5 days) to assess relative early and late gene expression. Cells were scraped and detached from culture dishes with cold PBS Ethylenediamine Tetra Acetic Acid (EDTA) solution, centrifuged, and incubated in RNAlater® (Thermo Scientific) solution. In short, the mRNA from the samples was amplified and converted into a library of cDNA fragments with attached adaptors. Each molecule was sequenced to generate Short Sequence Reads (SSRs) and the SSRs were aligned to a reference genome. A genomescale transcription map was produced and the total reads of each gene’s exons determined the quantifiable expression level of the gene. From the generated report, only genes that were significantly affected by GFF-B treatment with ≥ 2-fold up regulation or down regulation and concomitant p-values<0.05 (probability equation [45]) compared to the control were considered for further investigation, n=1.
Protein determination
Protein was extracted from cells using Radioimmunoprecipitation Assay (RIPA) buffer supplemented with protease and phosphatase inhibitors. Cells were centrifuged at 10,000 RPM for 10 minutes at 4°C. The protein supernatant/lysate was separated from the cell pellet and placed on ice. 2-10 μL aliquots were prepared with Pierce® BCA Protein Assay Kit (Thermo Scientific). The colorimetric assay was spectrophotometrically read at 570 nm absorbance with Bovine Serum Albumin (BSA) standard in a microplate reader (Bio-Rad, 550). Protein content results were calculated against the BSA standard curve and reported as μg of protein, n=3.
Western blot analysis
Whole protein lysates were extracted and separated from the cell pellet after incubation in RIPA-buffer as described above. Cytoplasmic and nuclear protein extraction was performed with NE-PER™. Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Scientific) according to the manufacturer’s instructions. Following harvest, lysate extraction, and protein determination, equal stock aliquots were frozen at -80°C until Western blot processing. Western blot analysis was performed with cell protein lysate employing analogous methods as previously described [41]. 40-80 μg protein aliquots were loaded onto 10% or 12% Sodium Dodecyl Sulfate Polyacrylamide Gels (SDS-PAGE) or 8-12% pre-cast midi gradient gels (Novex, Invitrogen), electrophoresed, and transferred to nitrocellulose membrane. Membranes were blocked with either 5% milk or BSA in Tris-buffered Saline Containing 1% Tween 20 (TBST). Primary antibodies include: HO1, NQO1, TXNRD1, and Nrf2 (Santa Cruz). The appropriate horseradish peroxidaseconjugated anti-rabbit or anti-mouse immunoglobin G secondary antibodies were applied (EMD Millipore). Actin HRP, and Lamin A/C were used as loading controls (Santa Cruz). Bands were detected by chemiluminescence with Molecular Imager VersaDoc (Bio-Rad, MP 5000) and then quantified by densitometry using ImageLab (Bio-Rad). Results are expressed as a percentage of the control. Numerical data is generated from small samples (plated at 1 × 106 cells) from three to four different cell lines, triplicates or quadruplicates of small samples from a single cell line, or large samples (plated at 3 × 106 cells) from one cell line, n=1-4.
Statistical analysis
Data was statistically analyzed using one-way ANOVA followed by F-Test and then Student’s t-test. Values were considered significant at p<0.05. Standard Deviation (SD) is reported where indicated.
Results
GFF preserves cell viability against induced cytotoxicity
The first experimental initiative was to determine the rescue potential of GFF for cell viability. We challenged NHEM with 4TBP, a phenolic depigmenting compound cytotoxic to melanocytes, alone or in combination with 0-10% GFF-B for 6 days. Cell viability was analyzed by MTT assay (Figure 1). 4TBP alone was the only treatment group statistically reduced in viability compared to the ethanol control. 5% GFF-B alone did not significantly alter viability and all concentrations of GFF-B, 2.5%, 5%, and 10%, rescued cell viability of the groups co-treated with 4TBP. This result demonstrated a protective effect against induced 4TBP cytotoxicity.
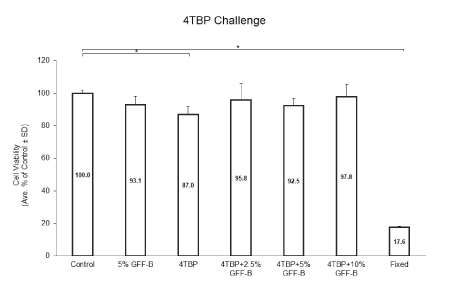
Figure 1: GFF effect on cell viability in 4TBP challenged NHEM.
NHEM were treated with or without 200 μM 4TBP alone or in combination with 2.5-10% GFF-B for 6 days. Cell viability was analyzed by MTT assay. Only 4TBP alone significantly diminished the cell viability compared to the ethanol control; thus, co-treatment with GFF-B at all concentrations demonstrated a protective effect in NHEM. Fixation was used as a negative control. Results are expressed as a percentage of the control. *p<0.05, SD, n=3.
GFF suppresses ROS generation
Next, we evaluated the antioxidant effect of GFF in our cell culture model using a fluorescent assay for ROS visualization by microscopy (Figure 2A). We challenged NHEM with 300 μM 4TBP in ethanol vehicle alone or in combination with 10% GFF-B for 90 minutes. Catalase, a known antioxidant that scavenges H2O2, was used as a positive control. As expected, 4TBP alone increased the fluorescent signal corresponding to ROS generation and was statistically increased above all other treatment groups. 4TBP and 10% GFF-B co-treatment resulted in a diminished fluorescent signal, suppressing the ROS induction. This result was equivalent to the weakened signal identified in the 10% GFF-B alone and the 4TBP and catalase co-treatment.
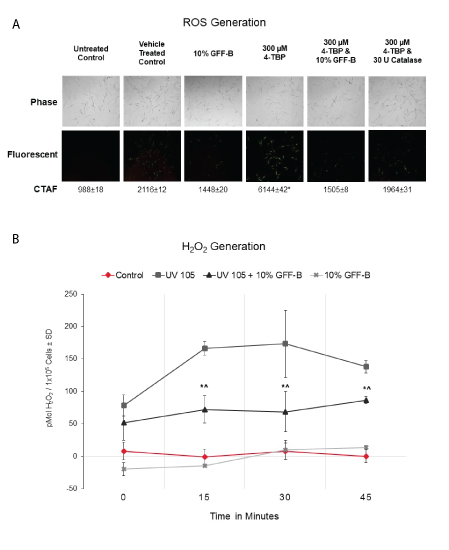
Figure 2: GFF effect on ROS generation induced by 4TBP and UVB.
(A) NHEM were challenged with 300 μM 4TBP alone or in combination with 10% GFF-B for 90 minutes. ROS were visualized with fluorescence microscopy and Corrected Total Area Fluorescence (CTAF) was determined using Image J. Co-treatment of GFF-B significantly suppressed ROS generation in NHEM exposed to 4TBP, an effect comparable to catalase-the positive control. Phase-contrast (above) and fluorescent (below) microscopy. *p<0.01, ± SD, n=30.
(B) NHEM dosed with 10% GFF-B and irradiated with UVB (105 mJ/cm2) were analyzed for H2O2 production until harvest at the indicated time points (0-45 minutes). GFF-B reduced H2 O2 generation in UVB irradiated NHEM at time points 15-45 minutes. Results are expressed in pMol H2O2 /1 × 105 cells, SD, n=3. *p<0.05 for UV 105+10% GFF-B compared to UV 105; ^p<0.05 for UV 105+10% GFF-B compared to Control.
We then performed a specific H2O2 quantification assay based on the fluorescence of luminol. Lightly pigmented NHEM were incubated with or without 10% GFF-B and irradiated with UVB 105 mJ/cm2 . The generation of H2O2 was then monitored over time (0, 15, 30, and 45 minutes) by assaying aliquots of the PBS in which the cells were kept (Figure 2B). At the 15-minute timepoint, 10% GFF-B significantly suppressed the UVB-induced H2O2 by 70% compared to the untreated and irradiated control. This suppression in H2O2 continued for the remainder of the experiment. Collectively, these results indicated that inhibition of ROS occurs in NHEM with GFF treatment, which ensures fast and sustained antioxidant effects.
GFF activates melanocyte antioxidant response
To understand the global gene expression changes induced by GFF treatment in human melanocytes, we procured Illumina HiSeq-based next generation sequencing RNA-seq analysis. From the generated report, we selected genes that were significantly affected by 10% GFF-B treatment at two timepoints (24 hours and 120 hours) with ≥ 2-fold upregulation or downregulation compared to untreated control cells for further investigation. RNA-seq analysis demonstrated a total of 3,574 genes altered by ≥ 2-fold and 533 of those genes had p-values <0.05 after 24 hours of GFF treatment. The second timepoint, 120 hours, revealed 3,466 genes were altered by ≥ 2-fold and 572 of the genes had p-values<0.05 due to GFF treatment (Figure S1). We focused our analysis on gene categories related to oxidative stress and endogenous antioxidant response, with initial attention on the 24 hours timepoint. Several gene targets associated with the adaptive oxidative stress response were modulated by ≥ 2-fold (Figure 3). Mitogen-Activated Protein Kinase 10 (MAPK10/JNK3) was upregulated while its potential downstream signaling target Jun B proto-oncogene (Jun B/AP-1) was downregulated indicating probable transcriptional activation of responsive stress pathways. Additionally, three antioxidant enzymes including Heme oxygenase 1 (HMOX1/HO1), NAD(P)H Quinone Oxidoreductase 1 (NQO1), and Thioredoxin Reductase 1 (TXNRD1) from the Nrf2-ARE pathway were significantly upregulated (Table S1). However, glutathione peroxidase 3 (GPX3) and Superoxide Dismutase 3 (SOD3), enzymes that neutralize H2O2 and superoxide anion (O2- ) respectively, were both downregulated. Downregulation of Cytochrome B-245, Alpha Polypeptide (CYBA), a protein involved in O2- production and phagocytosis, was also observed; signifying a protective output of ROS synthesis repression. This antioxidant response was partially maintained at the second timepoint 120 hours (5 days) (Table S2). In total, this data indicates that GFF is able to successfully activate antioxidant and protective gene transcription in NHEM.
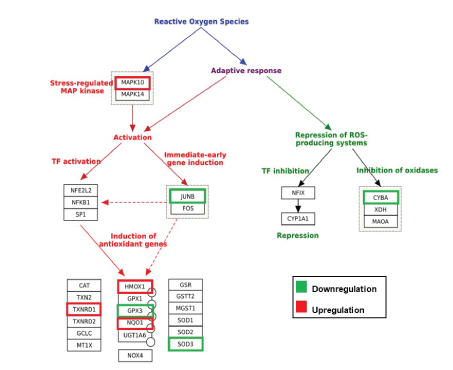
Figure 3: GFF effect on various oxidative stress targets in NHEM [Modified from Morel & Barouki, 1999].
RNA-seq transcriptome profiling analysis was performed with mRNA extracted from NHEM treated with 10% GFF-B for 24 hours and 120 hours (5 days). At the 24 hours time point, GFF-B treatment modulated several gene targets associated with oxidative stress at fold change ≥ 2, n=1; including MAPK10=Mitogen activated protein kinase 10; JUNB=Proto-oncogene; CYBA=Cytochrome B-245, Alpha polypeptide; HMOX1=Heme oxygenase 1; TXNRD1=Thioredoxin reductase 1; GPX3=Glutathione peroxidase 3; NQO1 =NAD(P)H Quinone oxidoreductase 1; SOD3=Superoxide dismutase 3, Extracellular. Green indicates downregulation, red indicates upregulation, and no color indicates fold change <2.
GFF upregulates Nrf2-ARE downstream targets HO1, NQO1, and TXNRD1
To validate the RNA-seq data, Western blot analysis was performed to elucidate the protein expression of the antioxidant targets. NHEM cultures were evaluated in both long- and short-term experimental protocols. In the long-term protocol, NHEM were treated with or without 10% concentration of GFF-A or GFF-B for 5-7 days. At the completion of dosing, whole cell protein lysates were extracted and analyzed (Figure 4A). NHEM treated with GFF-A and GFF-B showed significant increases of Nrf2 polyubiquitinated protein and the phase II enzymes HO1, NQO1, and TXNRD1 protein expression. Upregulation was relatively 2-fold in all experiments.
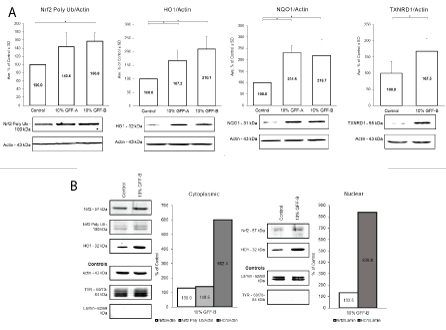
Figure 4: GFF effect on Nrf2-ARE pathway.
(A) NHEM were treated with or without 10% GFF-A and 10% GFF-B for 5 or 7 days. At harvest, protein expression was analyzed by Western blot and densitometry. Polyubiquitinated Nrf2 was increased by 44% and 57%, respectively. HO1 and NQO1 expression was nearly doubled by 10% GFF-A and 10% GFF-B. TXNRD1 was increased by 68% after treatment with 10% GFF-B. Results are expressed as a percentage of the control. *p <0.05, SD, n=3-4.
(B) NHEM were administered a single dose of 10% GFF-B for 8 hours. Cytoplasmic and nuclear fractions of the protein lysate were analyzed by Western blot and densitometry, n=1. Expression of Nrf2 was increased by 30% in the cytoplasm and the nucleus. Polyubiquitinated Nrf2 was also increased in cytoplasm by 41%. HO1 was highly inducible, showing 6-fold and 8-fold increases between the cytoplasm and nucleus. TYR and lamin were used as extraction separation controls where appropriate.
Short-term, single dose protocols were also performed with 10% GFF-B for investigation of separated cytoplasmic and nuclear protein lysate fractions. Initially, time course experiments were executed at 4, 8, and 24 hours to understand the trend of the protein expression and to select optimal timepoints (data not shown). Cytoplasmic and nuclear protein fractions from NHEM treated with 10% GFF-B for 8 hours were subsequently evaluated (Figures 4B,S2). Staining for TYR was done in the nuclear fraction (absence of TYR expression) and for lamin in the cytoplasmic fraction (absence of lamin expression) to confirm adequate separation of cytoplasmic and nuclear fractions. Increase in Nrf2 was demonstrated in both nuclear and cytoplasmic fractions of GFF treated NHEM compared to the controls. Notably, HO1 protein was increased 6- and 8-fold in both the cytoplasmic and nuclear fractions, respectively. This collection of experiments confirmed that GFF does upregulate the expression of antioxidant enzymes via activation of the Nrf2-ARE pathway. Although, Nrf2 was not significantly affected in the RNA-seq data, the adjustment to shorter timepoints in the Western blot experiments yielded positive results. We supplementally investigated the sustenance of GFF’s effect on Nrf2-ARE in NHEM after irradiation with UVB. NHEM were pretreated with or without 10% GFF-B for 6 days, irradiated with UVB 90 mJ/cm2 , and then treated again for an additional 24 hours. NQO1 and HO1 expression was evaluated at harvest by Western blotting (Figure S3). Analysis revealed NQO1 levels were maintained in NHEM post-irradiation. HO1 again demonstrated vast inducibility; however, expression was barely sustained over untreated, irradiated NHEM. Altogether, this data illustrates the positive ability of GFF to incite endogenous antioxidant capacity in NHEM through mediation of the Nrf2-ARE pathway.
Discussion
In this study, we demonstrate that GFF effectively suppresses ROS generation and successfully executes a protective capacity in NHEM assaulted with the cytotoxic, depigmenting compound 4TBP and with UVB irradiation. The data from our investigation leads us to conclude that GFF has effective antioxidant capacity in NHEM, which is accomplished in part by upregulation of antioxidant enzymes HO1, NQO1, and TXNRD1 through Nrf2-ARE pathway activation. The exact or earliest timing for Nrf2-ARE pathway transcription has not been elucidated yet. Results specifically from shorter timepoint (0-90 minutes) experiments also suggest that GFF may contain physiological components with non-enzymatic antioxidant action. There is most likely a collective of working mechanisms which include Nrf2-ARE to create the antioxidant protection response that requires additional study.
HO1 has indirect antioxidant activity that aids in the defense against ROS by catalyzing the degradation of heme into ferrous iron (Fe2+), CO, H2O, and biliverdin. Newly synthesized biliverdin is rapidly converted to bilirubin by Biliverdin Reductase (BVR) at the expense of NADPH. Bilirubin, a potent antioxidant, scavenges ROS primarily in lipophilic locations such as bilayer membranes [46-49]. Because of the high inducibility of HO1, the release of Fe2+ is a concern and could be associated with the elevated levels of ferritin reported in melanocytes compared to keratinocytes that correlated with increased DNA oxidative lesions [2]. Bioavailable iron is a source for cytotoxic Fenton reactions; therefore, more studies are needed to illustrate a clear picture of iron and heme homeostasis in melanocytes, especially after UVR exposure. HO1 levels were not maintained with GFF treatment in NHEM exposed to UVB, which could have been a protective response if the free iron becomes more threatening under irradiated conditions; however, UVA is more effective at inducing HO1 [50-52]. High ferritin levels matched with HO1 is considered protective by other authors [51,53]. Nevertheless, HO1 deficiency in various models does lead to oxidative stress and adverse pathologies [54-56].
NQO1 is primarily localized in the cytosol, existing as a homodimer with one molecule of FAD per monomer. It catalyzes single step 2-electron reductions of quinones using NADPH via a ping pong mechanism. This results in less reactive hydroquinone species and can yield substrates for phase II conjugation reactions that promote excretion; a strategy that would work well against the reactive quinone intermediates generated in melanin synthesis [57]. The protection from cytotoxicity induced by phenolic compound Rhododendrol (RD) via over expression of NQO1 in B16BL6 mouse melanoma and NHEM as described by [58] parallels to the GFF induced protection from 4TBP in our study. NQO1 additionally generates antioxidant forms of ubiquinone and Vitamin E and is proposed to have a role in O2- scavenging that would also serve well in this model [57,59].
TXNRD1 functions in the Thioredoxin (TXN) detoxification system throughout the cytoplasm. This system is led by Peroxiredoxin (PRDX), an antioxidant enzyme that reduces H2O2 and alkyl hydroperoxides by using thiols, primarily TXN, as electron donors in their catalytic action. TXNRD1 regenerates oxidized thioredoxin disulfide (TXN-S2) back to thioredoxin (TXN-(SH)2) using NADPH. This system plays essential roles in a variety of cellular functions including redox control of transcription factors, deoxyribonucleotide synthesis, and cell growth [60]. TXNRD1 is also capable of regenerating other antioxidant compounds such as ascorbic acid, selenium-containing substances, lipoic acid, and ubiquinone; and additionally supports α-tocopherol function [61].
GFF induced ARE responses have been reported in additional models. GFF co-treatment resulted in the downregulation of inducible Nitric Oxide Synthase (iNOS) expression and concurrent upregulation of HO1 expression in RAW264.7 mouse macrophages induced with Lipopolysaccharide (LPS) as documented by [39]. Potential modulation of the ARE pathway in human keratinocytes and fibroblasts occurred after comparing treatments with GFF and olive oil derivatives separately and in combination as described by Finlay DR, et al. [62]. HO1 expression was increased in a dose-dependent manner in both the primary cell cultures and human skin explant cultures when treated with GFF and/or olive oil derivatives. Yet, this group concluded that the ARE transcription demonstrated in ARE-32, a luciferasebased reporter cell line, was much lower than expected based on the magnitude of the HO1 upregulation observed in the primary cultures, leading them to suspect involvement of Hypoxia Inducible Factor 1 (HIF1), an alternative transcription factor that mediates HO1 [63]. HIF1 activation has not been linked with simultaneous upregulation of HO1, NQO1, and TXNRD1; thus, signifying an involvement of Nrf2- ARE. HO1 expression was also less affected by GFF in comparison to olive oil in this study. Individual and synergistic inhibition of ROS generation with GFF at 0.1% concentration and green tea flavonoid Epigallocatechin Gallate (EGCG) in human keratinocytes was reported by [64]. Each compound could not overcome TNFα induced ROS production alone; it was only effectively suppressed in combination. GFF may have a special utility in the therapy of oxidative driven dyspigmentation disorders like vitiligo. Vitiligo onset is typically instigated by increased facultative melanization that is commonly triggered by precipitating factors such as Ultraviolet Radiation (UVR), hormones, cytokines, trauma, etc. This induced melanization elevates cytotoxicity through increased melanin intermediates and ROS generation that is intolerable by the genetically predisposed vitiligo melanocyte, resulting in immune targeting and cell death [6]. GFF also has putative antimelanogenic capacity. The combined GFF antioxidant and anti-melanization properties would be useful in a prevention strategy against early onset vitiligo to potentially impede lesion progression, or as an accompaniment in depigmentation therapy. There is no cure currently available; hence, efficient prevention and remedial strategies that can halt the progression of oxidative stress in epidermal melanocytes are desirable. It is necessary that our initial investigation was completed with normal melanocytes with functional melanogenic components and metabolic processes to understand the impact of GFF in baseline conditions. Future studies of the molecular components of GFF to map specific ingredients with the effects observed in the melanocyte, particularly HO1 inducibility; and comparing our current data in normal melanocytes to GFF treatment in vitiligo melanocyte cultures will propel this objective forward.
Another noteworthy consideration is the common adversities associated with the use and manufacture of synthetic medications. A sector of the scientific community is recommitting to natural products research for pharmacological benefits. The production and manufacture of naturally derived and sustainably sourced treatments continues to rise, being well received by the public. Vitiligo patients also seek remedies involving natural compounds and methodology; yet, regulation, efficacy, and potency are still considerable challenges [65]. GFF is a filtered, yeast derived extract that exhibits comprehensive skin health benefits that may have the potential to address such impediments. Our results have an important positive impact towards developing GFF derived therapeutic agents that can prime the cellular environment for ROS assault, aiding in the prevention of oxidative driven disease and the sustainability of epidermal melanocytes in skin.
Author Contributions
Conceptualization, T.H. and R.E.B.; Methodology, J.K.W.C., A.K., A.L.K., and R.E.B.; Formal Analysis, J.K.W.C., A.K., A.L.K., and R.E.B.; Investigation, J.K.W.C., A.K., A.L.K., and R.E.B.; Resources, A.L.K., T.H., and R.E.B.; Data Curation, J.K.W.C., A.K., and R.E.B.; WritingOriginal Draft Preparation, J.K.W.C. and R.E.B.; Writing-Review & Editing, J.K.W.C., A.K., A.L.K., T.H., and R.E.B.; Visualization, J.K.W.C. and R.E.B.; Supervision, R.E.B.; Project Administration R.E.B..; Funding Acquisition, T.H. and R.E.B.
Funding
This research was supported by The Procter & Gamble Co. and the National Vitiligo Foundation, Inc.
Conflicts of Interest
The authors declare no conflicts of interest.
