Abstract
Focal segmental glomerulosclerosis (FSGS) is the most common primary glomerular disease in nephrotic patients in the United States, frequently leading to end stage renal disease (ESRD). The cellular variant is a rare form of FSGS commonly associated with poor outcome. We report a case of cellular variant FSGS with progressive kidney dysfunction successfully treated with plasma exchange (PE). A 49-year-old Caucasian female presented with two days of ankle edema and hypertension. Laboratory findings showed serum creatinine (SCr) 1.6 mg/dL, urine albumin/creatinine ratio (uACR) 2.8 g/g, haematuria 3+ and no immunological abnormalities. Kidney biopsy revealed a cellular FSGS variant with segmental endocapillary proliferation on light microscopic, negative immunofluorescence and widespread foot process effacement by electronic microscopic. Prednisolone 1 mg/Kg was started. Four days later the SCr worsened (3.6 mg/dL) and the patient became severely nephrotic with uACR of6.8g/g, quickly attaining a maximum of 24.6 g/g in a short time and albumin of 2.15g/dL. Pulsed methyl prednisolone was started. Despite a 10 course of steroids, no clinical improvement was observed. Considering the rapidly worsening renal function and severe nephrotic syndrome, PE was begun in association with mycophenolate mofetil and tacrolimus. Kidney function recovered after one week. Complete remission was achieved at 3rd week and remains in complete remission at 27 months follow-up. Prolonged remission is a challenge in primary FSGS. PE associated with combined immunosuppression was effective in the present case. The short and long-term effects of plasma exchange in primary FSGS should be evaluated in prospective studies.
Keywords
Focal Segmental Glomerulosclerosis; Cellular Variant; Plasmapheresis; Nephrotic Syndrome
Introduction
Focal segmental glomerulosclerosis (FSGS) is the most common diagnosis in adult nephrotic patients in the United States (35% off all cases), mainly in African American descents (50%) and is also the most common primary glomerular disease leading to end-stage renal disease (ESRD) in both Caucasian and African American descents. However, in a Spanish based population, membranous nephropathy (24%) is the leading cause of nephrotic syndrome in patients between 15 and 65 years, while FSGS is the 4th most common cause (12%) [1]. FSGS is characterized by podocyte injury and foot process effacement leading to the typical histologic findings of obliteration of glomerular capillaries by extracellular matrix accumulation, representing the focal and segmental sclerosis [2,3]. FSGS is a histological pattern that can represent different etiologies [4]. Based on the etiology, FSGS could be classified as idiopathic (primary), genetic, adaptive (secondary), virus-associated and drug-induced [2]. Primary or idiopathic FSGS is a ‘primary’ podocytopathy presumably caused by a putative circulating permeability factor and characterized by nephrotic syndrome and FSGS lesion at light microscopy with widespread foot process effacement in electron microscopy (EM) [4,2]. The successful treatment of recurrent FSGS after transplant with plasma exchange (PE) is in favor of a circulating permeability factor as a cause for this lesion pattern [5].
The histologic framework provided by the NY-Columbia classification describes five distinct FSGS variants based on light microscopy patterns [3], encompassing FSGS not otherwise specified (NOS), perihilar, cellular, tip lesion and collapsing variants. Cellular variant is a rare form of FSGS (3% of all FSGS) frequently presenting with severe nephrotic syndrome and associated with acute kidney injury and hypertension [6-8], commonly with poor outcomes [5]. We herein present a patient with cellular variant FSGS with severe nephrotic syndrome and rapid worsening of kidney functionsuccessfully treated with PE and immunosuppressive regimen.
Case Report
A 49-year-old Caucasian female with no relevant past medical history presented to her general practitioner with two days complain of ankle edema. The urine dipstick analysis showed 3+ protein. One week before, she had complained of abdominal pain and diarrhea for two days that spontaneously remitted. She had previous laboratory findings documenting normal kidney function and no family history of kidney disease. She also denied taking any drug recently. On observation at Nephrology Department showed blood pressure of 154/92 mmHg and 1+ pitting ankle edema. Laboratory findings showed serum creatinine (SCr) 1.6 mg/dL, urine albumin/creatinine ratio (uACR) 2.8 g/g, microscopic hematuria 3+, sediment with no acanthocytes, serum albumin 2.5 g/dL and total cholesterol 231 mg/dL. The chest X-ray showed bilateral pleural effusion and kidney ultrasound was unremarkable. Further investigation showed no complement abnormalities, negative autoantibodies and negative viral serology. A kidney biopsy was performed showing cellular FSGS variant, with exuberant segmental endocapillary proliferation with increased cell infiltration and slight mesangial expansion (Figure 1). There were no features of acute tubular injury or chronic interstitial nephritis lesions. Immunofluorescence was negative. Electron microscopy showed exuberant diffuse effacement of the pedicels, with no evidence of electron dense deposits or endothelial tubuloreticular inclusions (Figure 1). Treatment with steroids was started (prednisolone 1 mg/Kg/day) and she was discharged on loop diuretics and angiotensin converting enzyme inhibitor. Four days later, she was admitted with worsened hypertension (162/92 mmHg), anasarca and worsened kidney function (SCr 3.24 mg/d). On admission, she showed uACR of 6.8 g/g, attaining a maximum of 24.6 g/g in a short time and serum albumin was 2.15 mg/dL. Pulse methylprednisolone was done (0.5 g qd for 3 days). Considering the rapidly worsening kidney function with severe nephrotic proteinuria despite 10 days of steroid treatment, we changed the therapeutic approach. PE was initiated (6 daily and 1 on alternative day) associated to the previous dose of prednisolone (1 mg/Kg/day), mycophenolate mofetil (MMF) (1 g bid) and tacrolimus (0.5 mg bid, aiming for very low serum levels of 2 ng/mL). One week after this was started, she showed marked improvement with edema regression, normal blood pressure and recovering kidney function (eGFR 72 ml/min/1.72m2 ). Complete remission was achieved at 1 month follow-up (3rd week of treatment). High dose steroid was continued for another month and then slowly tapered off. MMF was also reduced at 7 month follow-up. At 15 month follow-up the patient remained in complete remission (Figure 2) and MMF was further reduced aiming for withdrawal. The patient relapsed two months later (uACR 3.6 g/g with normal kidney function). The MMF dose was then increased to 0.5 g bid and complete remission was achieved after 2 weeks. At 27 month followup, the patient remains in complete remission with no adverse events or side effects to report (Figure 2).
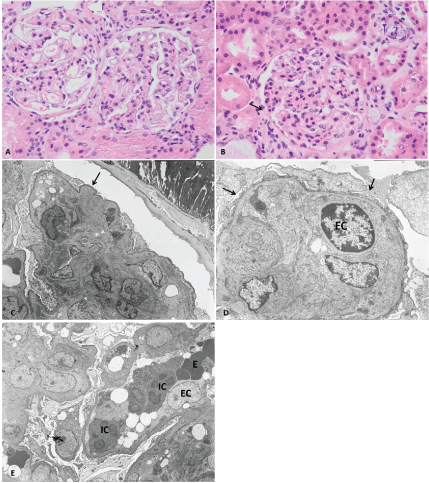
Figure 1: Renal biopsy light and electronic microscopy
Light microscopy: A– Exuberant cell proliferation with discrete mesangial expansion and increased cell infiltration by mononuclear; B – Segmental endocapillary hypercellularity, discrete mesangial expansion and apparent glomeruli-capsular synechiae (arrow). Electron microscopy: C– Area of segmental sclerosis (arrow); diffuse foot process effacemen; D – Capillary loop with exuberant hyperplasia of endothelial cells and diffuse foot process effacement (arrow); E – Capillary loop filled with cells (exuberant endocapillary proliferation), at the expense of leukocytes (inflammatory cell (IC)), erythrocytes (E), hyperplastic endothelial cells (EC) and platelets and diffuse foot process effacement.
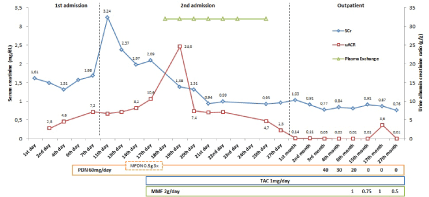
Figure 2: Renal function and proteinuria evolution during follow-up
Discussion
FSGS represents a histologic pattern of podocyte damage with different causes and frequently undefined pathogenesis. FSGS is one of the most serious glomerular diseases, with frequent progression to ESRD and with a high rate of recurrence in kidney allografts [4]. The clinical classification based on etiology distinguishes five forms: primary or idiopathic FSGS, the etiology of which is largely unknown; secondary or adaptive, which commonly refers to an adaptive response to glomerular hypertrophy/hyperfiltration or represent a nonspecific pattern of scarring due to a previous injury; genetic; drug-induced; virus-associated [2]. The mechanisms responsible for the podocytopathy are still unknown. A presumed circulating permeability factor has been proposed to play a role in the pathogenesis of primary FSGS [9]. Recurrence in renal allografts provides the best evidence for the existence of this circulating factor, but its nature remains unclear. The efficacy of PE in reducing proteinuria following recurrence reinforces this theory [5]. Additionally, there are diverse clinical and experimental studies that support the existence of circulating factors [4]. Despite extensive research, these circulating factors have not been identified or characterized. Five distinct FSGS variants were classified based on light microscopic patterns in NY-Columbia FSGS classification [3]: FSGS not otherwise specified (NOS), perihilar, cellular, tip lesion and collapsing variants. Several studies have documented significant differences in clinical findings and outcomes between these morphological patterns [6-7]. The cellular variant of FSGS is defined by the presence of at least one glomerulus with segmental expansion of the glomerular tuft with endocapillary hypercellularity, often with foam cells, with or without hyperplasia of overlying visceral epithelial cells and exclusion of FSGS tip and collapsing variants [3]. It usually presents with heavy proteinuria, severe nephrotic syndrome, acute kidney injury and hypertension in more than half of the cases [6-8]. It is less frequent then the other FSGS variants (3% of all FSGS [9]) and shows intermediate rates of remission (44.5%), acute kidney injury (57.1%) and progression to ESRD (27.8%), when compared to the collapsing and tip lesion variants [6]. Other authors have described 67% of cases with no remission [8].
However, the low number of cellular variant cases reported, make it difficult to understand its prognosis and response to treatment. Here we present a case of severe nephrotic syndrome and acute kidney injury secondary to cellular FSGS variant who was successfully treated with combined PE and immunosuppression, an approach similar to that used in allograft recurrent FSGS after kidney transplant and also described in therapy resistance FSGS in native kidney. This case shows a clinical presentation very close to what has been described in the literature. However, the patient kept getting worse after one week of steroid treatment. The notable worsening of kidney function and a significant increase in proteinuria led to a change to the treatment approach.
First-line treatment for primary FSGS is corticosteroids. Nevertheless, steroid resistance is common [4,10]. The identified predictors of kidney outcomes in FSGS are the amount of proteinuria, kidney function at presentation and tubule and interstitial damage. However, patients who achieve complete or partial remission have better renal outcomes than those who do not. Achieving complete or partial remission is the stronger predictor of kidney survival [11]. Therefore alternative immunosuppressive therapy is needed when steroids fail to achieve this goal. Calcineurin inhibitors, either cyclosporine or tacrolimus, can be used for steroid-resistant FSGS or with frequent relapses [10,11], particularly because these show an anti-proteinuric profile. MMF in association with low-dose prednisolone also seemed to be as effective as high-dose prednisolone in a prospective trial [12]. In our case, the rapid worsening of kidney function and the reported low remission rate in cellular FSGS variant led us to change the treatment for a combined immunosuppression. In order to rapidly control the disease progression while waiting for immunosuppression action, we believe that PE was crucial for the removal of a putative permeability factor and preventing further damage.
The presumed existence of a “glomerular permeability factor” explains the rapid relapse after transplantation and the efficacy of plasma exchange in this setting [4,3]. Plasma exchange has been used for treatment of transplant recurrence FSGS with suitable results [5]. In a systematic review and meta-analysis of PE treatment in post-transplant FSGS, overall complete or partial remission was 71% [13]. Rare cases have been published using this approach as rescue therapy in native kidney disease with encouraging results if used with concomitant immunosupression theraphy [13-15].
In a study including 11 patients with biopsy-proven FSGS, unresponsive to steroids and cytotoxic therapy, PE was performed 17 times during a median 22 weeks in addiction to oral prednisolone and intravenous cyclophosphamide. At 27 months follow-up 6 patients had complete remission (and stabilized kidney function) and 2 parcial remission [14].
On the other hand, less good results with PE were observed in others studies. In a small study including 5 patients with native kidney FSGS resistant to standard immunosuppressive therapy (steroids, cyclophosphamide and cyclosporine) and using immune adsorption, proteinuria reduction by more than 50% was observed in only 2 patients. In another study PE was performed in 8 patients with steroid-resistent idiopathic FSGS in native kidney, after a mean 12 months from initial presentation. These patients had received prednisone at a dose of 40 to 60 mg/day for an average of 4.4 months and in 5 this was followed by three 1g doses of IV methylprednisolone. Six PE treatments were then performed over 2 weeks and most patients continued to receive a tapering dose of prednisone. Proteinuria decreased in two of eight patients, although only transiently in one of the two. Both had stable renal function in the last follow-up [15]. The lack of greater response observed in these 2 last studies could be related to less intensive immunosuppression therapy.
In our case, early PE with combined immunosuppression showed effective to achieve complete remission. It also showed good results in long term follow-up with normal kidney function and no side effects of treatment. Since cellular variant FSGS is associated with just one third of response to steroids and rapid kidney loss, PE can be an option for preventing further damage in severe cases and long-term morbidity improving clinical outcome. The independent effect of PE in the treatment of primary FSGS in native kidney disease has yet be confirmed in prospective studies as well as its long-term effect.
Conflict of interest
none to declare
Article Information
Article Type: Case Report
Citation: Cunha L, Pereira F, Manso R, Fervenza F, Soto K (2017) Cellular Variant of Focal Segmental Glomerulosclerosis Treated with Plasma Exchange. Int J Nephrol Kidney Failure 3(2): doi http://dx.doi.org/10.16966/2380-5498.146
Copyright: © 2017 Cunha L, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original *Corresponding author: author and source are credited.
Publication history:
Received date: 16 Sep 2017
Accepted date: 04 Oct 2017
Published date: 10 Oct 2017